Clear Sky Science · ru
Жизнеспособная мышиная модель RIPK3 без киназной активности D143N выявляет её каркасную функцию в запуске воспалительных нарушений, индуцированных TNF
Почему это исследование на мышах важно для понимания воспаления
Многие тяжёлые заболевания — от смертельно опасных инфекций до обострений аутоиммунных расстройств — вызваны не только микроорганизмами или генетикой, но и саморазвивающимся воспалением. Белок RIPK3 долгое время считали ключевым исполнителем жестокой формы гибели клеток, подпитывающей такое воспаление, что делало его привлекательной целью для лекарств. Однако у RIPK3 есть и другие, менее изученные функции внутри клетки. В этом исследовании описана новая лабораторная мышиная модель, которая чётко разделяет киназную (убийственную) активность RIPK3 и его сигнальную «каркасную» роль, показывая вклад каждой в воспаление и указывая на новые терапевтические стратегии.
Два способа, которыми белок смерти может действовать
Клетки могут умирать упорядоченно или беспорядочно. При аккуратной, «безмолвной» гибели клетки перерабатываются без сильной тревоги. При более хаотичной форме — некроптозе — клетки разрываются и выливают содержимое, вызывая мощные иммунные реакции. RIPK3 является центральным звеном некроптоза: при активации он запускает другой белок, который проделывает отверстия в мембране клетки. В то же время предыдущие работы предполагали, что RIPK3 может помогать запускать классическую, каспаз-зависимую апоптозу и усиливать воспалительные сигналы даже без гибели клеток. Разделить эти роли было трудно, потому что существующие инактивированные варианты RIPK3 либо приводили к гибели эмбрионов, либо заметно понижали уровень белка, что мешало изучению его нормальной каркасной функции.
Более безопасный способ отключить убийственную функцию
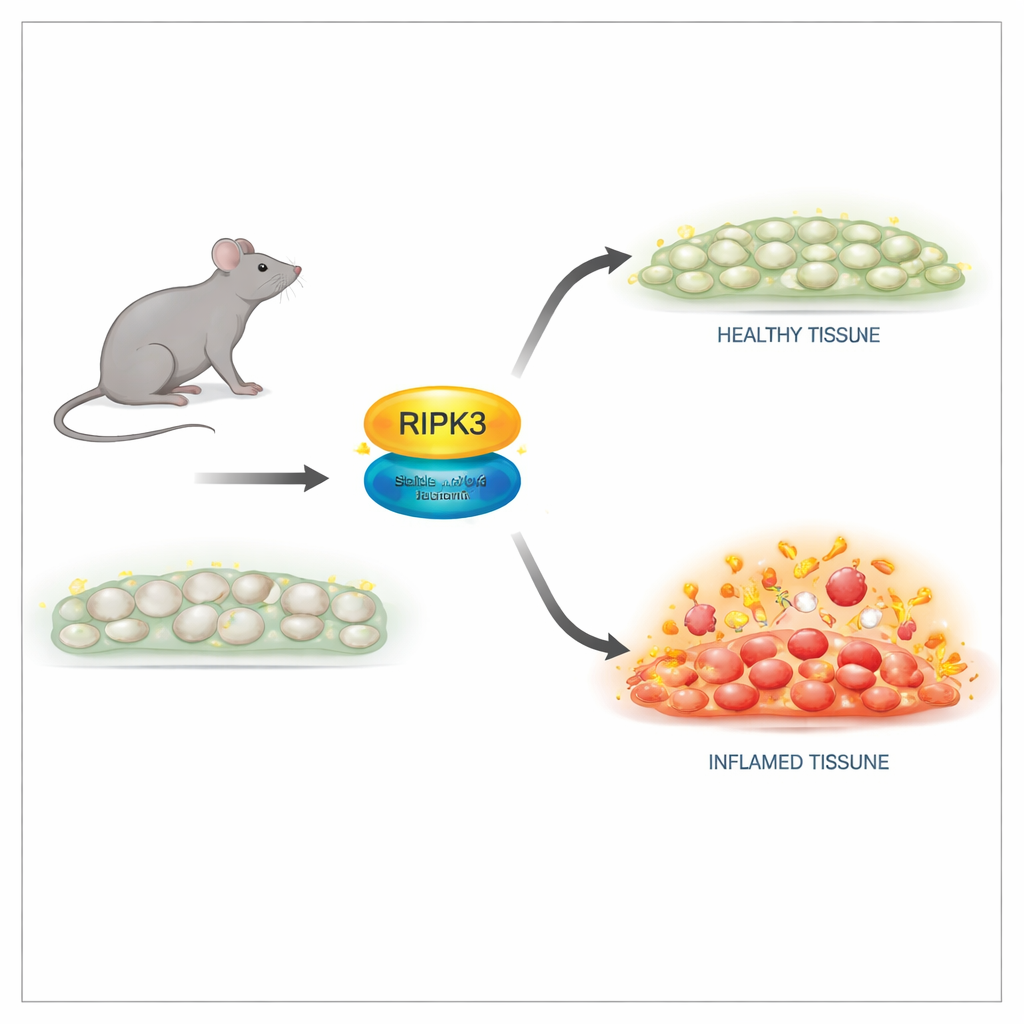
Исследователи создали мышей с тонкой заменой в белке RIPK3 в одной позиции, обозначенной как D143N, которая выключает ферментативную активность, сохраняя при этом структуру белка. В клетках этих животных уровни RIPK3 и архитектура тканей выглядели нормальными, а сами животные рождались и развивались так же, как здоровые сибсы. Важно, что клетки с версией D143N оказались полностью устойчивыми к множественным триггерам некроптоза, включая сигналы от фактора некроза опухоли (TNF), толл-подобных рецепторов и вирусных инфекций. Мутантный RIPK3 больше не мог активировать своего взаимодействующего партнёра или формировать разрушительный комплекс, необходимый для разрыва мембраны, но при этом он не провоцировал самопроизвольный апоптоз, избегая летальных побочных эффектов, наблюдаемых у старых мутантов RIPK3.
Разделение развития и болезни
Одна из наиболее известных ролей RIPK3 проявляется в эмбрионах, лишённых другого ключевого белка — каспазы-8: без каспазы-8 некроптоз, управляемый RIPK3, убивает эмбрион. В этом исследовании введение версии D143N RIPK3 полностью спасало таких обычно нежизнеспособных мышей. Они нормально развивались и были плодовиты, что доказывает: киназная (убийственная) активность RIPK3 не обязательна для нормального развития, если сохраняется его структура. Однако при введении взрослым мышам высоких доз TNF для индуцирования шокоподобного воспалительного синдрома картина изменилась. Животные, полностью лишённые RIPK3, были надёжно защищены от гибели, повреждения тканей и повышения уровня воспалительных молекул в крови. Мыши с версией D143N, несмотря на отсутствие некроптоза, были защищены лишь частично. Это указывает на то, что неклеточная, каркасная роль RIPK3 по‑прежнему способствует развитию воспаления.
Каркасная сигнализация, поддувающая жар
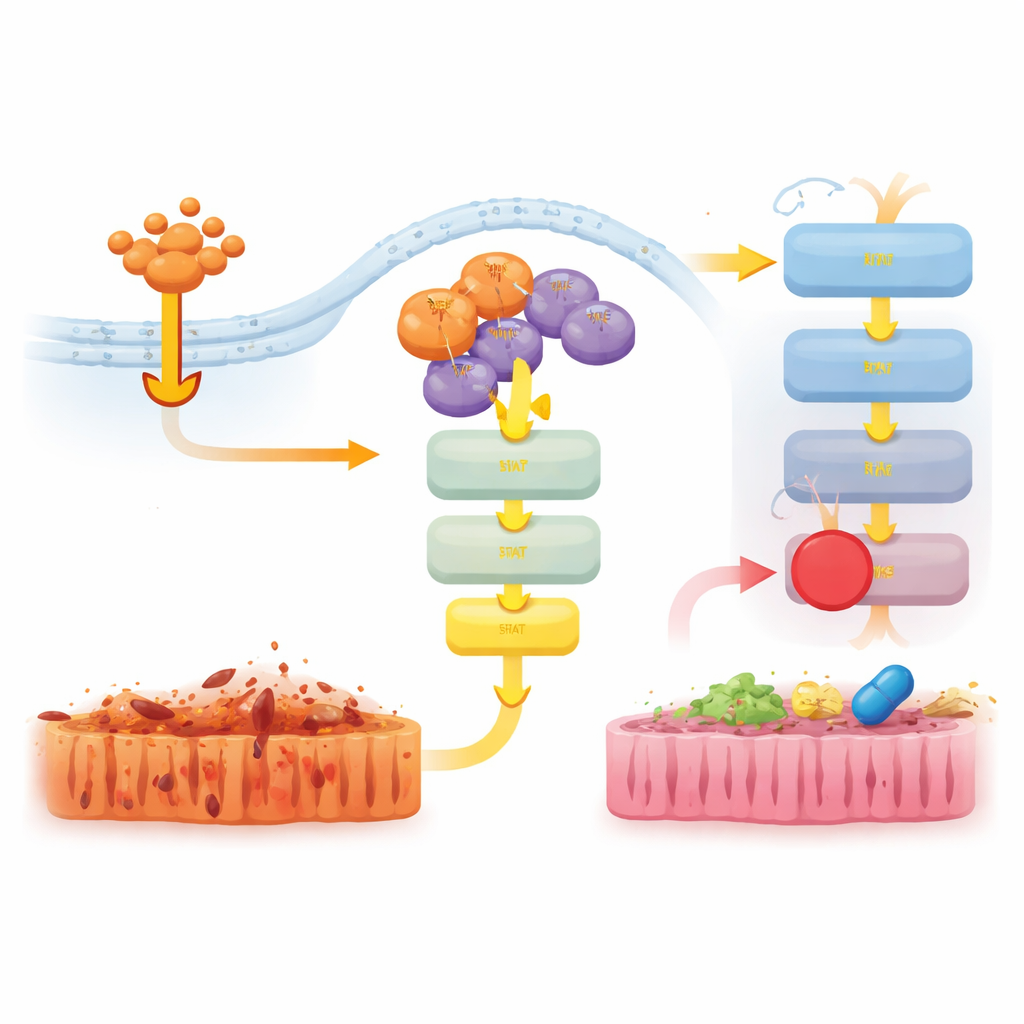
Чтобы понять этот нелекарственный вклад, исследователи изучили активность генов в кишечнике мышей, обработанных TNF. У животных без RIPK3 многие воспалительные гены были сильно подавлены. У мышей D143N подавление было слабее, и гены, связанные с интерферонным и врождённым иммунитетом, оставались более активными. На уровне белков TNF мощно включал сигнальные пути JAK–STAT1 и ERK у нормальных и D143N мышей, тогда как при полном удалении RIPK3 эта активация была почти полностью утрачена. Это показало, что даже без своей киназной функции физическое присутствие RIPK3 в сигнальных комплексах помогает передавать TNF‑сигналы в профлоговое воспалительное программирование через JAK–STAT1.
Ослабление вредных сигналов с помощью целевых препаратов
Затем исследователи проверили, может ли блокирование этих нижестоящих путей смягчить болезнь у D143N мышей при TNF‑индуцированном шоке. Лечение животных ингибитором JAK1/2, но не ингибитором ERK, уменьшало потерю температуры тела, снижало уровень провоспалительной молекулы IL‑6 и уменьшало повреждение тканей кишечника и гибель клеток. Отдельный ингибитор, нацеленный на другой белок — RIPK1, также серьёзно защищал мышей и подавлял активацию JAK–STAT1 и ERK. В совокупности результаты указывают на то, что каркасная функция RIPK3 действует совместно с RIPK1 для активации JAK–STAT1 и запуска воспаления, и что прерывание этой сигнальной цепочки может уменьшить повреждение тканей даже при уже блокированном некроптозе.
Что это значит для будущих терапий
В течение многих лет RIPK3 рассматривали главным образом как переключатель токсичной формы гибели клеток, и разработка лекарств сосредотачивалась на подавлении его ферментативной активности. Это исследование показывает, что этого может быть недостаточно: RIPK3 по‑прежнему может выступать как физическая платформа, усиливающая воспалительные сигналы через JAK–STAT1, способствуя шоку и повреждению тканей. Новая мышиная модель D143N наглядно выявляет эти двойственные роли, предоставляя мощный инструмент для изучения того, когда и как каждая функция имеет значение в различных заболеваниях. Для пациентов работа предполагает, что сочетание препаратов, нацеленных на RIPK3 или RIPK1, с блокаторами JAK–STAT1 может быть более эффективным способом подавления вредного воспаления при состояниях, вызванных TNF и связанными цитокинами.
Цитирование: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
Ключевые слова: RIPK3, некроптоз, воспаление, шок, индуцированный TNF, JAK-STAT1