Clear Sky Science · pl
In silico typowanie mapuje naturalną różnorodność transporterowo-zależnych otoczek Escherichia coli
Dlaczego cukrowa powłoka bakterii ma znaczenie
Wiele szczepów Escherichia coli, powszechnego bakterium jelitowego i częstej przyczyny poważnych zakażeń, pokrytych jest na powierzchni cukrową otoczką. Ta śliska powłoka pomaga im unikać układu odpornościowego i przetrwać w różnych gospodarzy oraz środowiskach. Przez dekady naukowcy mieli trudności z klasyfikacją i śledzeniem tych otoczek, ponieważ tradycyjne testy laboratoryjne były powolne i często zawodnie. Badanie pokazuje, jak nowoczesna analiza DNA może zmapować pełną różnorodność tych otoczek, ujawniając pominięte typy i pomagając w projektowaniu przyszłych szczepionek oraz terapii celowanych.
Od probówek do typowania komputerowego
Wcześniejsze prace klasyfikowały otoczki E. coli przy użyciu przeciwciał rozpoznających struktury powierzchniowe, procesu znanego jako serotypowanie. Testy te były pracochłonne, niedokładne i szczególnie trudne w przypadku otoczek, które mogą naśladować ludzkie cząsteczki i wywoływać słabe odpowiedzi odpornościowe. W rezultacie typowanie otoczek w dużej mierze zanikło pod koniec XX wieku, a jedynie podzbiór znanych typów otoczek był dobrze zbadany. W międzyczasie sekwencjonowanie genomów stało się tanie i powszechne, ale brakowało kompletnego odniesienia łączącego DNA otoczek ze znanymi typami. Luka ta uniemożliwiała badaczom wiarygodne rozpoznawanie nowych wariantów otoczek i zrozumienie ich rozmieszczenia wśród pacjentów, zwierząt i środowiska.
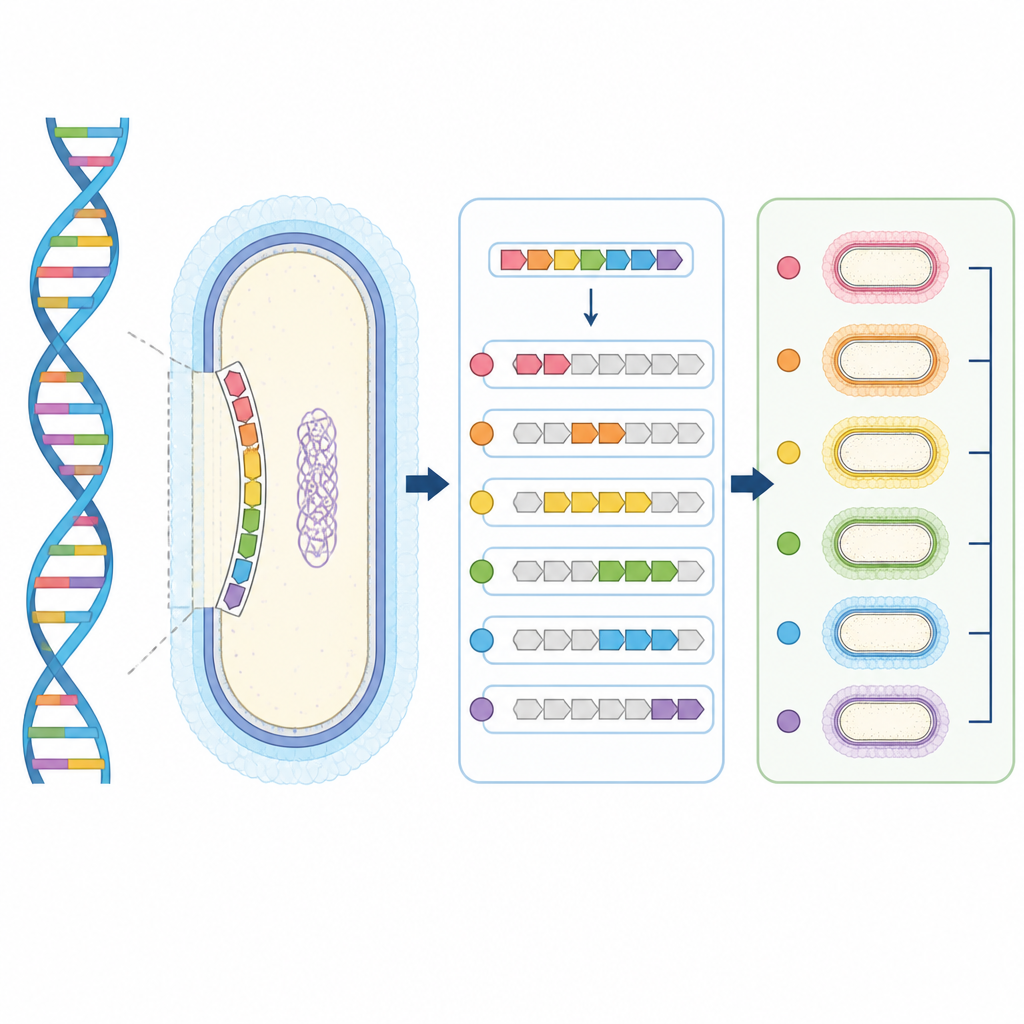
Budowanie genetycznej atlasu otoczek E. coli
Autorzy skupili się na głównej grupie otoczek E. coli, które zależą od systemu transportowego do przenoszenia cukrowej powłoki na powierzchnię komórki. Najpierw sekwencjonowali historyczną kolekcję szczepów referencyjnych, których otoczki zostały wcześniej zdefiniowane metodami klasycznymi. Poprzez dopasowanie struktur otoczek do ich podstawowego DNA stworzyli jasną mapę od genotypu do serotypu dla 35 ustalonych transporterowo-zależnych otoczek, którą skonsolidowali do 30 genetycznie odrębnych typów. Następnie przeanalizowali ponad 37 000 publicznie dostępnych genomów E. coli. Korzystając z kluczowego genu otoczki jako punktu orientacyjnego, wydzielili otaczające regiony DNA i pogrupowali je w unikatowe locus otoczkowe na podstawie wspólnej zawartości genów.
Odkrywanie nowych rodzin i funkcji otoczek
To wielkoskalowe przeszukanie ujawniło 85 odrębnych transporterowo-zależnych typów otoczek, w tym 55, które nie były częścią pierwotnej kolekcji referencyjnej. Analizując wspólne geny rdzeniowe budujące i eksportujące otoczkę, zespół podzielił te locus na cztery linie genetyczne, a nawet zidentyfikował wcześniej nierozpoznaną podgrupę. Aby zrozumieć, jakie struktury te otoczki mogą tworzyć, połączyli przeszukiwania domen, przewidywanie struktur białek i porównania z znanymi rodzinami enzymów. Podejście to pozwoliło przypisać prawdopodobne funkcje ponad 90 procentom genów specyficznych dla otoczek. W niektórych przypadkach zastosowano spektrometrię mas na oczyszczonych otoczkach, aby rozwiązać niezgodności między przewidywanymi genami a starszymi opisami chemicznymi, aktualizując proponowaną strukturę niektórych typów otoczek.
Nowe narzędzie do odczytu typów otoczek z genomów
Mając ten katalog, badacze opracowali kTYPr, narzędzie programowe, które czyta sekwencje genomowe i przewiduje typ otoczki. Zamiast polegać na prostych dopasowaniach sekwencji, kTYPr używa ukrytych modeli Markowa, które uchwytują wzorce w ramach rodzin białek i tolerują naturalne zmienności. Narzędzie najpierw sprawdza obecność podstawowych genów otoczki, a następnie ocenia, który specyficzny zestaw enzymów otoczkowych najlepiej pasuje do danego genomu. Strategia ta potrafi rozróżnić blisko spokrewnione otoczki, rozpoznać przestawione klastry genów oraz obsłużyć niekompletne genomy złożone z próbek metagenomicznych.
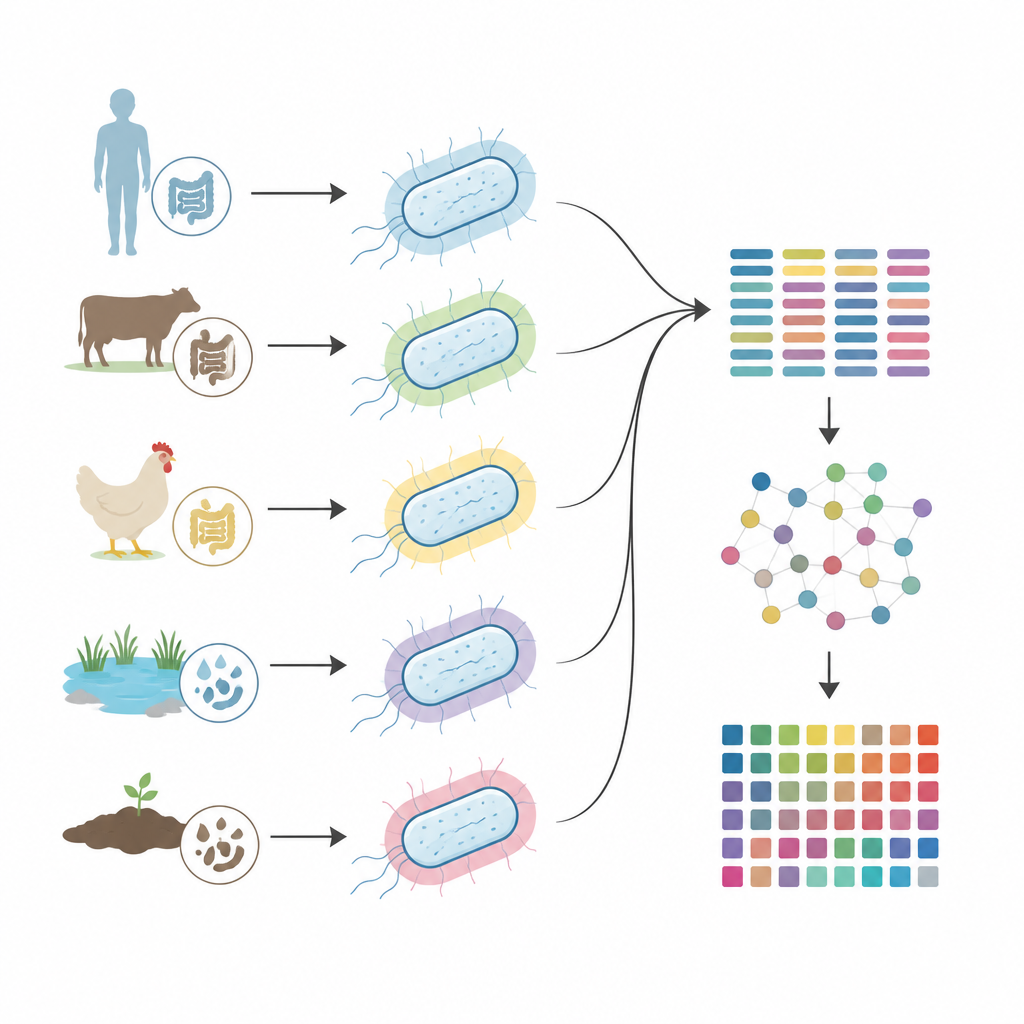
Różnorodność otoczek wśród gospodarzy, siedlisk i chorób
Zespół zastosował kTYPr do ponad 24 000 starannie wyselekcjonowanych genomów E. coli pochodzących od ludzi, zwierząt, żywności i źródeł środowiskowych, a także do prawie 3 000 fragmentów genomów zrekonstruowanych ze stolca zdrowych osób. Stwierdzili, że około jedna czwarta wszystkich genomów nosiła kompletny locus transporterowo-zależnej otoczki, a takie otoczki były szczególnie częste w szczepach pochodzących od ludzi, zwierząt domowych i środowisk powiązanych z działalnością człowieka. Nowe, wcześniej nieopisane typy otoczek były wzbogacone w mniej zbadanych środowiskach, takich jak zwierzęta dzikie, hodowlane i żywność. U ludzi te same typy otoczek występowały zarówno w zdrowych społecznościach jelitowych, jak i w szczepach powodujących zakażenia dróg moczowych, bakteriemię i zapalenie opon mózgowo-rdzeniowych, choć niektóre typy otoczek były silniej powiązane z chorobami inwazyjnymi niż inne.
Co to oznacza dla kontroli i zapobiegania zakażeniom
Dzięki stworzeniu szczegółowej mapy od genów otoczek do typów otoczek i udostępnieniu tego w przyjaznym oprogramowaniu, badanie przekształca niegdysiejszą zagadkową cukrową powłokę E. coli w coś, co można rutynowo śledzić w danych genomowych. Praca ujawnia znacznie większą różnorodność otoczek niż wcześniej sądzono i pokazuje, że wiele typów związanych z chorobami jest także powszechnych w zdrowym jelicie, gdzie mogą występować jako cisi kolonizatorzy, czasem powodując ciężkie zakażenia. Ten nowy genetyczny atlas i zestaw narzędzi pomogą badaczom badać, jak otoczki kształtują ekologię E. coli, jak wchodzą w interakcje z układem odpornościowym i fagami oraz jak można je precyzyjniej targetować w przyszłych szczepionkach i terapiach.
Cytowanie: Miravet-Verde, S., Cacace, E., Mores, C.R. et al. In silico typing maps the natural diversity of Escherichia coli transporter-dependent capsules. Nat Microbiol 11, 1217–1232 (2026). https://doi.org/10.1038/s41564-026-02323-5
Słowa kluczowe: Escherichia coli, otoczki bakteryjne, typowanie genomowe, różnorodność mikrobowa, cele szczepionek