Clear Sky Science · pl
Hamowanie HSP27 aktywuje interakcję XBP1s/CerS1, co wywołuje DRP1‑zależną mitofagię, chroniąc przed śmiercią komórki i sprzyjając cyklowi litycznemu KSHV w komórkach chłoniaka wysiękowego
Gdy odpowiedź na stres staje się dwusiecznym mieczem
Nasze komórki przetrwają codzienne urazy, włączając programy awaryjne, które naprawiają uszkodzenia i utrzymują je przy życiu. Komórki nowotworowe potrafią jednak zawłaszczyć te same mechanizmy, by się rozwijać i ukrywać w nich wirusy. Artykuł opisuje, jak zahamowanie pojedynczego białka chroniącego przed stresem w rzadkim chłoniaku nie tylko popycha komórki nowotworowe ku śmierci, lecz także daje ukrytemu w nich wirusowi okno do przebudzenia i replikacji. Zrozumienie tej delikatnej równowagi może pomóc w opracowaniu terapii, które zabijają nowotwór, jednocześnie uniemożliwiając wirusowi rozprzestrzenianie się.
Ukryty wirus w agresywnym chłoniaku
Chłoniak wysiękowy to wysoce agresywny nowotwór komórek B, rodzaju białych krwinek. Większość komórek tego guza nosi uśpionego pasażera: związany z mięsakami Kaposiego wirus herpes (KSHV). W stanie cichym, czyli latentnym, wirus produkuje niewiele białek i ukrywa się w genomie gospodarza. Pewne stresy mogą wypchnąć go w aktywną, lityczną fazę, w której się replikuje i tworzy nowe cząstki wirusowe, zwykle prowadząc do śmierci komórki gospodarza. Same komórki nowotworowe zależą od kilku systemów odpowiedzi na stres, w tym tzw. białek szoku cieplnego i odpowiedzi na nieprawidłowo sfałdowane białka (UPR), które pomagają im radzić sobie z nieprawidłowo sfałdowanymi białkami, zaburzeniami metabolizmu lipidów oraz uszkodzeniami mitochondriów produkujących energię. 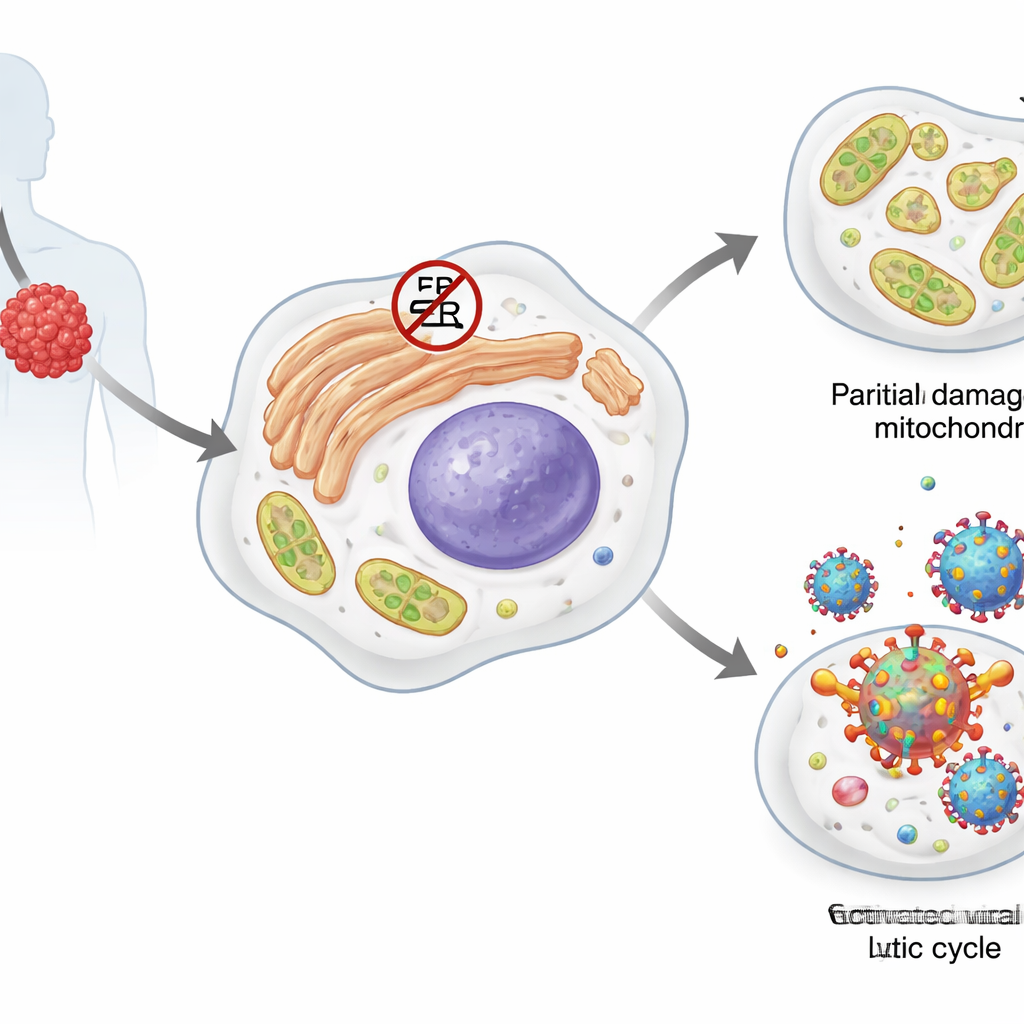
Blokowanie komórkowego ochroniarza
Naukowcy skoncentrowali się na HSP27, małym białku szoku cieplnego znanym z ochrony komórek przed stresem. Używając chemicznego inhibitora J2 lub uciszając gen za pomocą małych RNA, zmniejszyli aktywność HSP27 w komórkach chłoniaka hodowanych in vitro. Sprawiło to, że komórki były mniej zdolne do przetrwania i wywołało silny sygnał stresowy w wewnętrznej sieci błonowej zwanej retikulum endoplazmatycznym. Markery tej odpowiedzi, w tym czynniki ochronne i pro‑apoptotyczne, wzrosły, a kluczowy przełącznik zwany XBP1s został uruchomiony. Równocześnie komórki wykazywały więcej oznak programowanej śmierci, co potwierdza, że usunięcie HSP27 popycha je ku punktowi krytycznemu między przetrwaniem a zagładą.
Obieg stresu, który komunikuje się z lipidami komórkowymi
Stres w retikulum endoplazmatycznym jest ściśle powiązany z tym, jak komórki gospodarują lipidami. Zespół odkrył, że blokada HSP27 zwiększa poziomy CerS1, enzymu syntetyzującego specyficzny lipid — C18‑ceramid. Gdy chemicznie zablokowali XBP1s, wzrost CerS1 zniknął, co pokazuje, że XBP1s pomaga włączać gen CerS1 w tych warunkach. Co uderzające, hamowanie CerS1 obniżało z kolei poziomy XBP1s, ujawniając dodatnie sprzężenie zwrotne: każdy z tych czynników wspiera drugi. Ta molekularna interakcja nie tylko przekształca metabolizm lipidów, lecz także wzmacnia zdolność komórki do adaptacji na stres retikulum endoplazmatycznego, nawet gdy sygnały prowadzące do śmierci narastają.
Mitochondria recyklingowane zamiast natychmiast niszczone
Stres w jednej części komórki często przelewa się na mitochondria, maleńkie elektrownie wytwarzające energię. Po zahamowaniu HSP27 komórki chłoniaka wytwarzały więcej reaktywnych form tlenu, co świadczy o kłopotach mitochondrialnych, i zwiększyły poziom DRP1, białka rozdzielającego mitochondria na mniejsze fragmenty. Autorzy pokazali, że pętla XBP1s–CerS1 odpowiadała za wzrost DRP1. To z kolei wywołało mitofagię, proces kontroli jakości, w którym uszkodzone mitochondria są otaczane pęcherzykami i dostarczane do „centrów recyklingu” komórkowego — lizosomów. Przy użyciu barwników fluorescencyjnych i markerów białkowych potwierdzili selektywne usuwanie mitochondriów. Gdy chemicznie lub genetycznie zablokowali DRP1, ta mitofagia zmalała, a komórki umierały chętniej, co oznacza, że recykling mitochondriów faktycznie pomagał zestresowanym komórkom nowotworowym przetrwać. 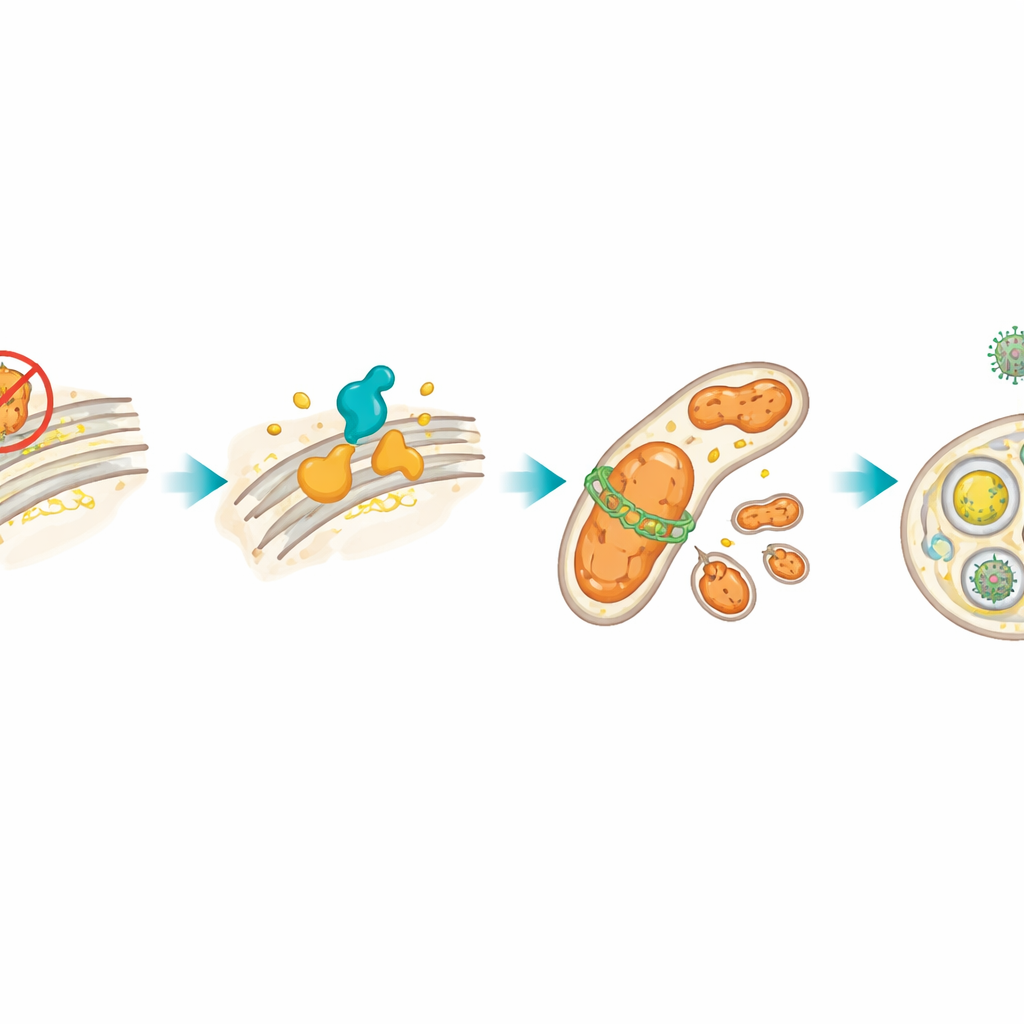
Dając wirusowi czas na ucieczkę
Ta sama mitofagia, która chroniła komórki guza, sprzyjała też KSHV. Aktywacja XBP1s, akumulacja C18‑ceramidu i zwiększona fragmentacja mitochondriów zostały powiązane z przebudzeniem tego wirusa. Tutaj, po zahamowaniu HSP27, więcej komórek wykazywało ekspresję wczesnych i późnych białek wirusowych, co jest wyraźnym znakiem replikacji litycznej. Zablokowanie DRP1, a zatem mitofagii, zmniejszyło tę reaktywację wirusa. Autorzy sugerują, że przez nieznaczne wydłużenie przeżycia komórki pod stresem, mitofagia daje KSHV czas na ukończenie cyklu replikacyjnego i opuszczenie umierającej komórki, potencjalnie zakażając nowe cele i przyczyniając się do rozwoju nowotworu.
Co to oznacza dla przyszłych terapii
Dla czytelnika niebędącego specjalistą kluczowy przekaz jest taki, że HSP27 działa jako centralny koordynator ruchu drogowego dla sposobu, w jaki komórki chłoniaka radzą sobie ze stresem, jak recyklingują uszkodzone mitochondria i kiedy wirus związany z nowotworem decyduje się obudzić. Wyłączenie HSP27 uruchamia łańcuch zdarzeń, który zarówno osłabia przeżywalność komórki, jak i — paradoksalnie — tymczasowo ją chroni poprzez mitofagię, jednocześnie umożliwiając replikację KSHV. W ujęciu terapeutycznym połączenie hamowania HSP27 z lekami blokującymi DRP1‑zależną mitofagię mogłoby szybciej doprowadzać do śmierci komórek guza i ograniczać szansę wirusa na rozprzestrzenienie się, oferując dwutorową strategię przeciwko temu śmiertelnemu chłoniakowi.
Cytowanie: Gonnella, R., Corrado, V., Scaffidi, G.F. et al. Inhibiting HSP27 activates the XBP1s/CerS1 interplay, which triggers DRP1-driven mitophagy, thereby protecting against cell death and promoting the KSHV lytic cycle in primary effusion lymphoma cells. Cell Death Discov. 12, 118 (2026). https://doi.org/10.1038/s41420-026-02979-2
Słowa kluczowe: chłoniak wysiękowy, wirus związany z mięsakami Kaposiego, odpowiedź na stres komórkowy, mitofagia, białko szoku cieplnego HSP27