Clear Sky Science · pl
Dehydrogenaza acylo-CoA krótkołańcuchowa inicjuje demetylację mtDNA i jego wyciek, zasilając odporność przeciwnowotworową w raku jelita grubego
Dlaczego nasze własne komórki czasem ukrywają nowotwór przed układem odpornościowym
Rak jelita grubego jest jednym z najbardziej śmiertelnych nowotworów na świecie, częściowo dlatego, że obrona immunologiczna często nie rozpoznaje go i nie atakuje. To badanie odkrywa nieoczekiwane powiązanie między tym, jak komórki nowotworowe spalają tłuszcze, jak ich małe elektrownie (mitochondria) obchodzą się z własnym DNA, a tym, czy układ odpornościowy zostaje zaalarmowany o obecności guza. Śledząc ten łańcuch zdarzeń, autorzy wyróżniają też znany naturalny związek — hipericynę — jako potencjalny sposób ponownego wzbudzenia ataku odpornościowego w raku jelita grubego.
Brakujący „strażnik” mitochondrialny w guzach jelita
Zespół rozpoczął od przeszukania dużych zestawów danych z człowieka i myszy, by znaleźć geny metaboliczne, które konsekwentnie zmieniają się w raku jelita grubego. Jedne z odgrywających rolę enzymów wyróżniły się: dehydrogenaza acylo-CoA krótkołańcuchowa, znana jako ACADS, która normalnie pomaga mitochondriom rozkładać krótkołańcuchowe kwasy tłuszczowe. W próbkach pacjentów i w kilku modelach mysich poziomy ACADS były wyraźnie niższe w tkance guzowej niż w pobliskim zdrowym jelicie. Gdy naukowcy zmniejszyli ekspresję ACADS w mysich komórkach raka jelita, guzy rosły szybciej i bardziej agresywnie; zwiększenie ACADS spowalniało wzrost guza. Myszy zablokowane genetycznie w kierunku braku ACADS specyficznie w wyściółce jelitowej rozwinęły więcej i większe guzy w chemicznym modelu raka związanego z zapaleniem, co wspiera tezę, że ACADS działa jako supresor nowotworowy w jelicie.
Jak guzy wyciszają sygnały alarmowe odporności
Te efekty wzrostu nie dały się wytłumaczyć tylko szybkością podziałów komórek nowotworowych in vitro, która zmieniła się niewiele. Zamiast tego utrata ACADS napędzała wzrost guza jedynie u zwierząt z nienaruszonym układem odpornościowym, co wskazuje na zmiany w mikrośrodowisku guza. Analizy pojedynczych komórek ludzkich raków jelita grubego wykazały, że guzy o niskim poziomie ACADS są otoczone większą liczbą komórek nowotworowych i komórek immunosupresyjnych — takich jak mieloidowo‑pochodne komórki supresorowe, pewne makrofagi i limfocyty T regulatorowe — oraz mniejszą liczbą pomocnych komórek T i komórek NK. Ten wzorzec wskazuje na „immunosupresyjne sąsiedztwo”, które chroni nowotwór przed atakiem. 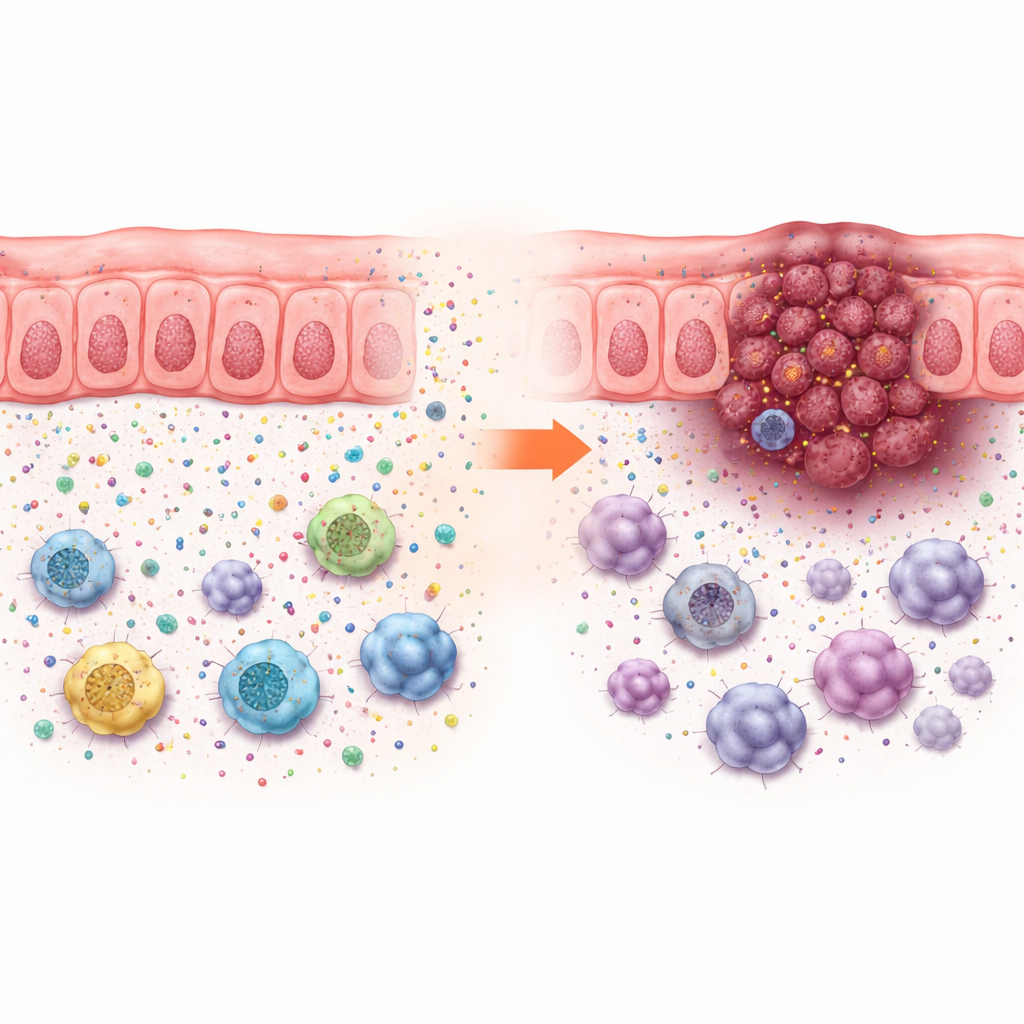
Wyciek DNA mitochondrialnego jako ukryty wyzwalacz
Co łączy enzym spalający tłuszcze z czujnikiem DNA? Odpowiedź tkwi w mitochondrialnym DNA (mtDNA). Pod wpływem stresu fragmenty mtDNA mogą uciekać z mitochondriów do płynu wewnątrzkomórkowego, gdzie cGAS rozpoznaje je jako sygnał niebezpieczeństwa. Badacze wykazali, że komórki nowotworowe pozbawione ACADS miały mniej mtDNA w tym przedziale, mimo że całkowita zawartość mtDNA nie uległa zmianie. Zablokowanie wycieku mtDNA w komórkach o wysokim ACADS wyłączyło cGAS–STING, potwierdzając, że te uwolnione fragmenty DNA są krytycznym alarmem. Co zaskakujące, klasyczne czynniki stresu mitochondrialnego, takie jak reaktywne formy tlenu, skoki wapnia czy duże zmiany kształtu mitochondriów, nie wyjaśniały w pełni różnic. Zamiast tego badanie wskazuje na „bramy” w błonie mitochondrialnej i, co ważniejsze, na chemiczne znaczniki na samym mtDNA. 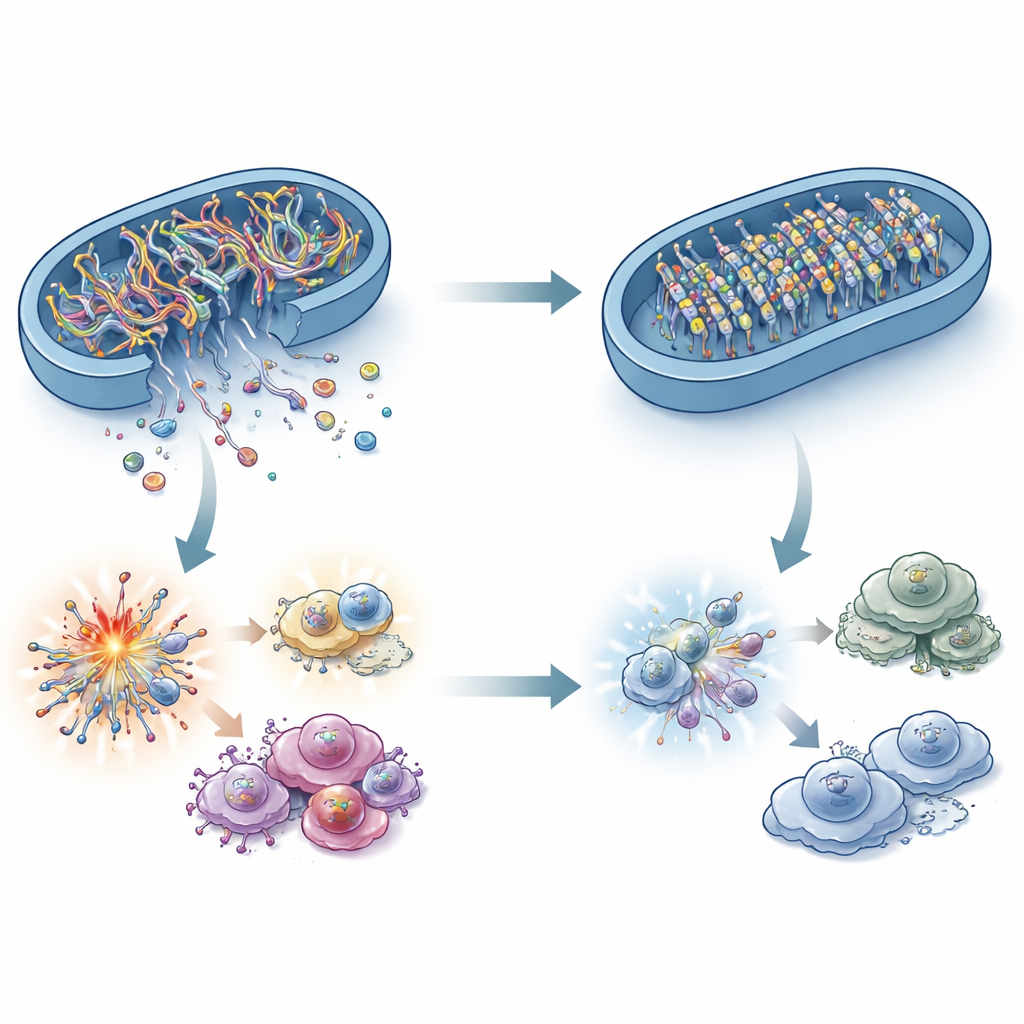
Partner metylujący DNA, który zamyka alarm
Poprzez skryning interakcji białek wykryto, że ACADS wiąże się z formą enzymu metylującego DNA, DNMT1, która lokalizuje się w mitochondriach. Gdy ACADS zniknęło, mitochondrialna forma DNMT1 się kumulowała, dokładając dodatkowe grupy metylowe na mtDNA. Te znaczniki czynią mtDNA bardziej stabilnym i mniej podatnym na pękanie i wyciek. Nadmiar mitochondrialnego DNMT1 zmniejszał ucieczkę mtDNA, tłumił sygnalizację cGAS–STING i przyspieszał rozwój guza, podczas gdy blokada DNMT1 lekiem dekitabina przywracała wyciek mtDNA i spowalniała guzy pozbawione ACADS. Próbki od pacjentów odzwierciedlały te obserwacje: niski poziom ACADS korelował z wysokim mitochondrialnym DNMT1, słabszą sygnalizacją STING, mniejszą liczbą efektorowych komórek T, większą liczbą komórek supresyjnych i gorszym przewidywanym odzewem na immunoterapię z użyciem inhibitorów punktów kontrolnych.
Przywracanie odporności związkiem z dawnych badań
Aby sprawdzić, czy tę ścieżkę można wykorzystać terapeutycznie, badacze przeprowadzili komputerowy przeszuk związków wiążących ACADS. Zidentyfikowali hipericynę, naturalny barwnik wcześniej testowany jako terapia aktywowana światłem w niektórych chłoniakach skórnych. W komórkach raka jelita grubego hipericyna zwiększała poziomy ACADS, redukowała mitochondrialny DNMT1, zwiększała wyciek mtDNA i reaktywowała sygnalizację cGAS–STING — zmiany zależne od obecności ACADS. W modelach mysich i w krótkotrwałych hodowlach ludzkich guzów jelita grubego leczenie hipericyną zmniejszało rozmiary guzów lub przesuwało skład komórek odpornościowych w kierunku bardziej aktywnego stanu bogatego w komórki T. Chociaż potrzebne są dalsze badania przed zastosowaniem klinicznym, wyniki sugerują, że farmakologiczne „włączenie” ACADS może pomóc przekształcić zimny, immunosupresyjny guz w taki, który lepiej odpowiada na immunoterapię.
Co to znaczy dla pacjentów i przyszłych terapii
Mówiąc prościej, praca ta pokazuje, że niektóre raki jelita grubego rosną częściowo dlatego, że wyciszają mitochondrialny enzym, który normalnie pomaga uwalniać maleńkie fragmenty DNA do wnętrza komórki, gdzie działają jak sygnały świetlne przywołujące układ odpornościowy. Pozwalając partnerowi metylującemu DNA zablokować to mitochondrialne DNA, guzy cierpiące na niedobór ACADS ukrywają te sygnały i unikają wykrycia przez odporność. Przywrócenie aktywności ACADS, na przykład za pomocą leków podobnych do hipericyny, mogłoby ponownie otworzyć ten mitochondrialny system alarmowy, wzmocnić odporność przeciwnowotworową i poprawić odpowiedzi na istniejące immunoterapie. W związku z tym ACADS, mitochondrialny DNMT1 i aktywność szlaku STING mogą służyć jako użyteczne biomarkery i cele w dążeniu do skuteczniejszych terapii raka jelita grubego.
Cytowanie: Yang, F., Wang, M., Hu, S. et al. Short-chain acyl-CoA dehydrogenase initiates mtDNA demethylation and leakage to fuel antitumor immunity in colorectal cancer. Sig Transduct Target Ther 11, 113 (2026). https://doi.org/10.1038/s41392-026-02675-8
Słowa kluczowe: rak jelita grubego, odporność przeciwnowotworowa, DNA mitochondrialne, metabolizm lipidów, szlak cGAS-STING