Clear Sky Science · fr
Un modèle murin viable RIPK3 inactif en kinases D143N révèle sa fonction de échafaudage dans l’induction d’un trouble inflammatoire par le TNF
Pourquoi cette étude chez la souris est importante pour l’inflammation
De nombreuses maladies sévères, des infections mortelles aux poussées auto-immunes, sont alimentées non seulement par des germes ou des gènes, mais par une inflammation incontrôlée de l’organisme lui‑même. Une protéine appelée RIPK3 est depuis longtemps considérée comme un acteur central d’une forme violente de mort cellulaire qui alimente cette inflammation, ce qui en fait une cible médicamenteuse attractive. Mais RIPK3 a aussi d’autres rôles intracellulaires, moins bien compris. Cette étude décrit un nouveau type de souris de laboratoire qui sépare proprement l’activité tueuse de RIPK3 de son rôle de « support » signalétique, révélant comment chacun contribue à l’inflammation et ouvrant la voie à de nouvelles stratégies thérapeutiques.
Deux manières d’agir pour une protéine de mort
Les cellules peuvent mourir de façon ordonnée ou désordonnée. Dans la mort cellulaire « silencieuse » et ordonnée, l’organisme recycle discrètement les éléments cellulaires sans alerter fortement le système immunitaire. Dans une forme plus chaotique appelée nécroptose, les cellules éclatent et répandent leur contenu, déclenchant de fortes réponses immunitaires. RIPK3 est au centre de la nécroptose : une fois activée, elle met en marche une autre protéine qui perce des trous dans la membrane cellulaire. Cependant, des travaux antérieurs ont suggéré que RIPK3 peut aussi favoriser la mort cellulaire classique pilotée par les caspases et stimuler des voies inflammatoires même sans tuer les cellules. Distinguer ces rôles a été difficile car les formes inactives existantes de RIPK3 provoquaient soit la mort embryonnaire, soit une chute marquée du niveau de protéine, rendant l’étude de son comportement d’échafaudage difficile.
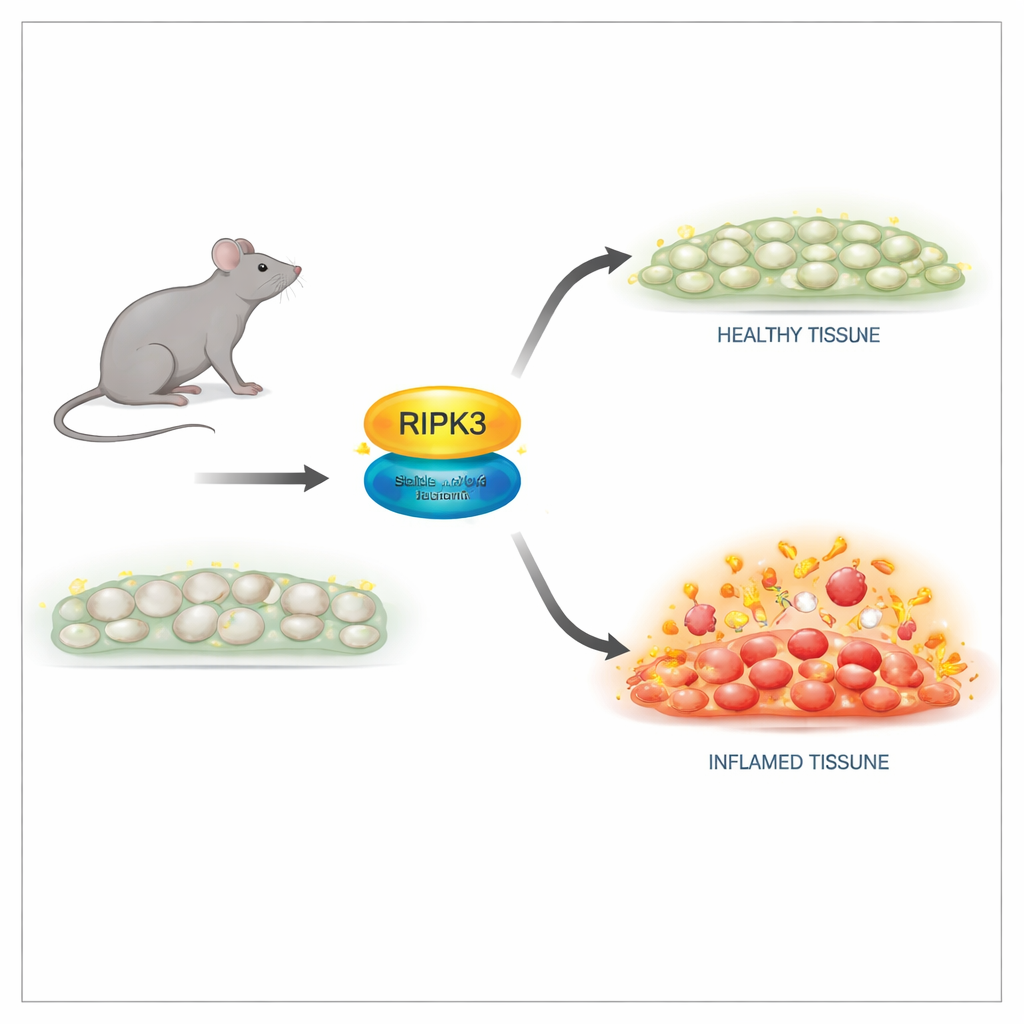
Une façon plus sûre d’éteindre la fonction tueuse
Les chercheurs ont conçu des souris portant une modification subtile de la protéine RIPK3 en un seul site, appelée D143N, qui élimine son activité enzymatique tout en préservant sa structure. Dans les cellules issues de ces souris, les niveaux de RIPK3 et l’architecture tissulaire semblaient normaux, et les animaux naissaient et se développaient comme leurs congénères sains. De manière importante, les cellules portant la version D143N étaient totalement résistantes à de multiples déclencheurs de nécroptose, y compris les signaux du facteur de nécrose tumorale (TNF), des récepteurs de type toll et des infections virales. Le RIPK3 mutant ne pouvait plus activer son partenaire en aval ni former le complexe destructeur nécessaire à la rupture membranaire, tout en n’induisant pas d’apoptose spontanée, évitant ainsi les effets létaux observés avec d’anciens mutants de RIPK3.
Séparer le développement de la maladie
Un des rôles les mieux connus de RIPK3 concerne les embryons dépourvus d’une autre protéine clé, la caspase‑8 : sans caspase‑8, la nécroptose déclenchée par RIPK3 tue l’embryon. Dans cette étude, l’introduction de la version D143N de RIPK3 a complètement sauvé ces souris autrement non viables. Elles se sont développées normalement et étaient fertiles, prouvant que l’activité tueuse de RIPK3 est dispensable pour le développement normal lorsqu’on conserve sa structure. Pour autant, chez les souris adultes exposées à une forte dose de TNF pour induire un syndrome inflammatoire de type choc, le tableau a changé. Les animaux dépourvus totalement de RIPK3 étaient fortement protégés contre la mort, les lésions tissulaires et les molécules inflammatoires dans le sang. Les souris porteuses de D143N, malgré l’absence de nécroptose, n’étaient que partiellement protégées. Cela indique que le rôle non‑tueur, d’échafaudage, de RIPK3 contribue encore à promouvoir l’inflammation.
Un signal d’échafaudage qui attise les flammes
Pour comprendre cette contribution non létale, l’équipe a examiné l’activité génique dans l’intestin de souris traitées au TNF. Chez les animaux déficients en RIPK3, de nombreux gènes inflammatoires étaient fortement atténués. Chez les souris D143N, cependant, cette suppression était moins marquée, et les gènes liés aux réponses aux interférons et à l’inné restaient plus actifs. Au niveau protéique, le TNF a fortement activé les voies de signalisation JAK–STAT1 et ERK chez les souris normales et D143N, mais cette activation était presque entièrement absente lorsque RIPK3 était supprimée totalement. Cela montre que, même sans sa fonction tueuse, la présence physique de RIPK3 dans des complexes de signalisation aide à transmettre les signaux du TNF vers un programme pro‑inflammatoire via JAK–STAT1.
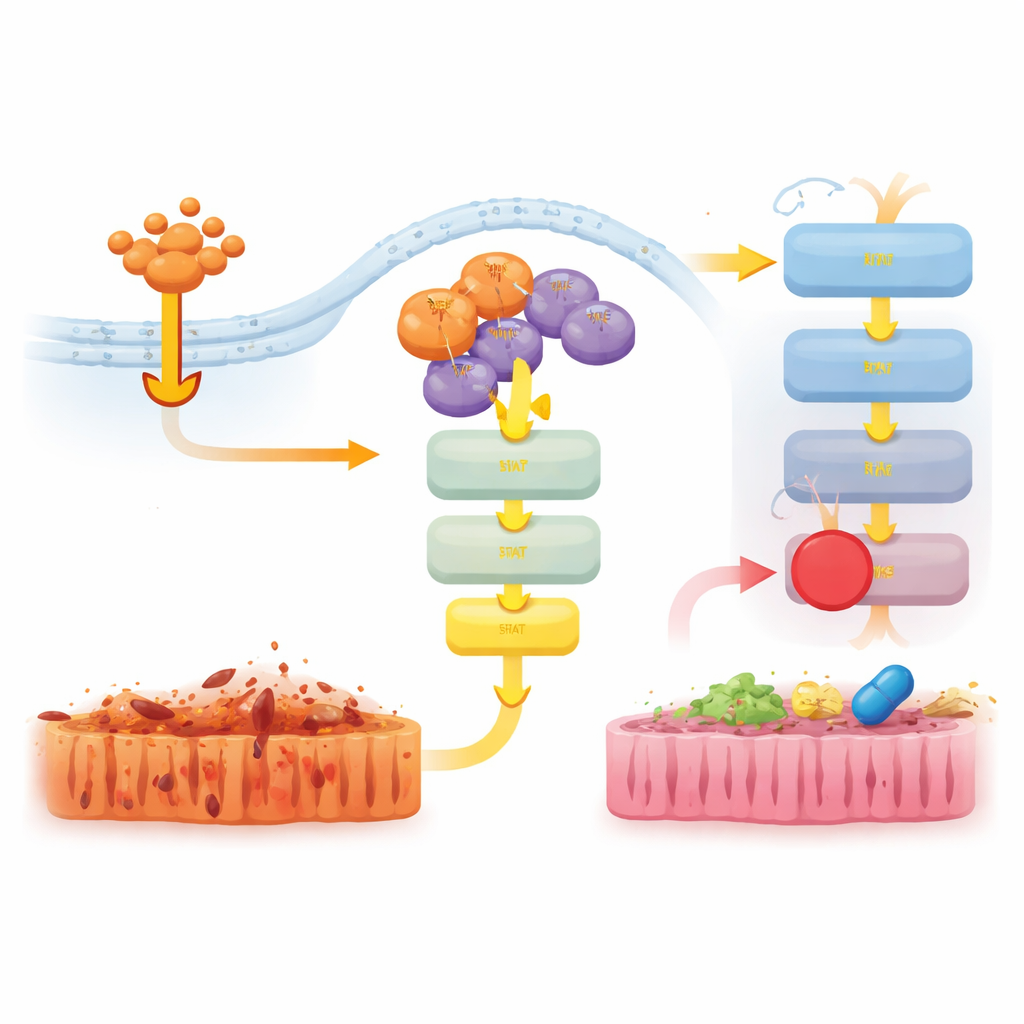
Atténuer les signaux nocifs avec des médicaments ciblés
Les chercheurs ont ensuite testé si le blocage de ces voies en aval pouvait atténuer la maladie chez les souris D143N soumises au choc induit par le TNF. Le traitement des animaux par un inhibiteur de JAK1/2, mais pas par un inhibiteur d’ERK, a réduit la perte de température corporelle, fait baisser les niveaux de la cytokine inflammatoire IL‑6 et diminué les lésions et la mort cellulaire dans l’intestin. Un inhibiteur distinct ciblant une autre protéine, RIPK1, a également fortement protégé les souris et atténué l’activation de JAK–STAT1 et ERK. Ensemble, ces résultats suggèrent que la fonction d’échafaudage de RIPK3 fait équipe avec RIPK1 pour activer JAK–STAT1 et conduire l’inflammation, et que l’interruption de cette signalisation peut réduire les lésions tissulaires même lorsque la nécroptose est déjà bloquée.
Ce que cela signifie pour les traitements futurs
Pendant des années, RIPK3 a été essentiellement perçu comme l’interrupteur d’une forme toxique de mort cellulaire, et le développement de médicaments s’est concentré sur l’inhibition de son activité enzymatique. Cette étude montre que cela peut ne pas suffire : RIPK3 peut encore agir comme une plate‑forme physique qui amplifie les signaux inflammatoires via JAK–STAT1, contribuant au choc et aux lésions tissulaires. Le nouveau modèle murin D143N révèle ces rôles doubles avec une clarté inhabituelle, établissant un outil puissant pour étudier quand et comment chaque fonction compte dans différentes maladies. Pour les patients, ces travaux suggèrent que combiner des médicaments ciblant RIPK3 ou RIPK1 avec des bloqueurs de JAK–STAT1 pourrait calmer plus efficacement l’inflammation nocive dans des affections drivées par le TNF et des cytokines apparentées.
Citation: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
Mots-clés: RIPK3, nécroptose, inflammation, choc TNF, JAK-STAT1