Clear Sky Science · es
Análisis integrador de predicciones in silico y evidencia clínica para delinear la capacidad de la secuenciación HiFi de lectura larga en genes paralogados
Por qué importa desenredar los genes que se parecen
Nuestro ADN contiene muchos pares o familias de genes “gemelos” que se parecen casi de forma idéntica. Estos fragmentos copiados y pegados del código son importantes para la salud y la enfermedad, pero son notoriamente difíciles de leer correctamente con las pruebas genómicas estándar. Este estudio plantea una pregunta práctica para la medicina: ¿hasta qué punto puede la secuenciación de lectura larga actual separar realmente estas copias confusas de genes, en qué puntos todavía falla y puede el software inteligente ayudar a cerrar las brechas restantes?
Cuando las copias de genes engañan a las pruebas de ADN convencionales
Las pruebas genéticas modernas a menudo se basan en fragmentos cortos de ADN, de solo unos pocos cientos de letras. Cuando esas lecturas cortas provienen de regiones donde las copias de genes son casi idénticas, un ordenador no puede saber fácilmente a cuál copia pertenecen. Esto puede difuminar o esconder cambios importantes relacionados con enfermedades. Para capturar este problema de una manera que no dependa de un solo paciente o equipo, los autores usaron un concepto llamado “mapeabilidad”: para una longitud de lectura elegida, preguntaron con qué frecuencia esa secuencia exacta aparece en el genoma de referencia. Si una lectura podía coincidir en varios lugares, esa región se marcaba como difícil o imposible de resolver con lecturas cortas.
Lo que las lecturas de ADN más largas pueden arreglar —y lo que no pueden
El equipo primero mapeó qué partes del genoma serían problemáticas para la secuenciación de lectura corta utilizada de forma general y encontró que 645 genes de importancia médica caían en esta zona de riesgo. Luego repitieron el cálculo para lecturas mucho más largas, de hasta 14.000 letras de ADN, similar a lo que producen hoy las tecnologías de lectura larga de vanguardia. Con estas lecturas largas, aproximadamente dos tercios de los genes previamente problemáticos se predijo que pasarían a ser claramente legibles, pero aproximadamente un tercio permaneció obstinadamente irresuelto. Cuando compararon estas predicciones con datos clínicos reales de lectura larga procedentes de 66 personas, los genes predichos como “arreglables” mostraron, de hecho, mapeos de alta confianza con mucha más frecuencia que aquellos predichos como difíciles, confirmando que las simulaciones capturaron el comportamiento del mundo real.
Comprobando la precisión en el mundo real, no solo la teoría
Los investigadores fueron más allá de las puntuaciones de mapeabilidad y examinaron directamente qué tan bien podían detectarse las variantes genéticas en las regiones problemáticas. Usando un genoma de referencia bien estudiado, demostraron que la secuenciación de lectura larga halló tanto cambios de una sola letra como pequeñas inserciones/deleciones más completamente que los métodos de lectura corta en áreas conocidas por ser difíciles de mapear. Las lecturas cortas no detectaron muchos cambios verdaderos en estas regiones, mientras que las lecturas largas captaron casi todos ellos, aunque siguieron produciendo llamadas algo más inciertas que en las partes fáciles del genoma. Al modelar cómo disminuye el número de genes no resueltos a medida que aumenta la longitud de lectura, encontraron una curva que baja rápidamente hasta unas 7–8 mil letras y luego se aplana, lo que sugiere que simplemente hacer las lecturas aún más largas no eliminará todos los puntos ciegos.
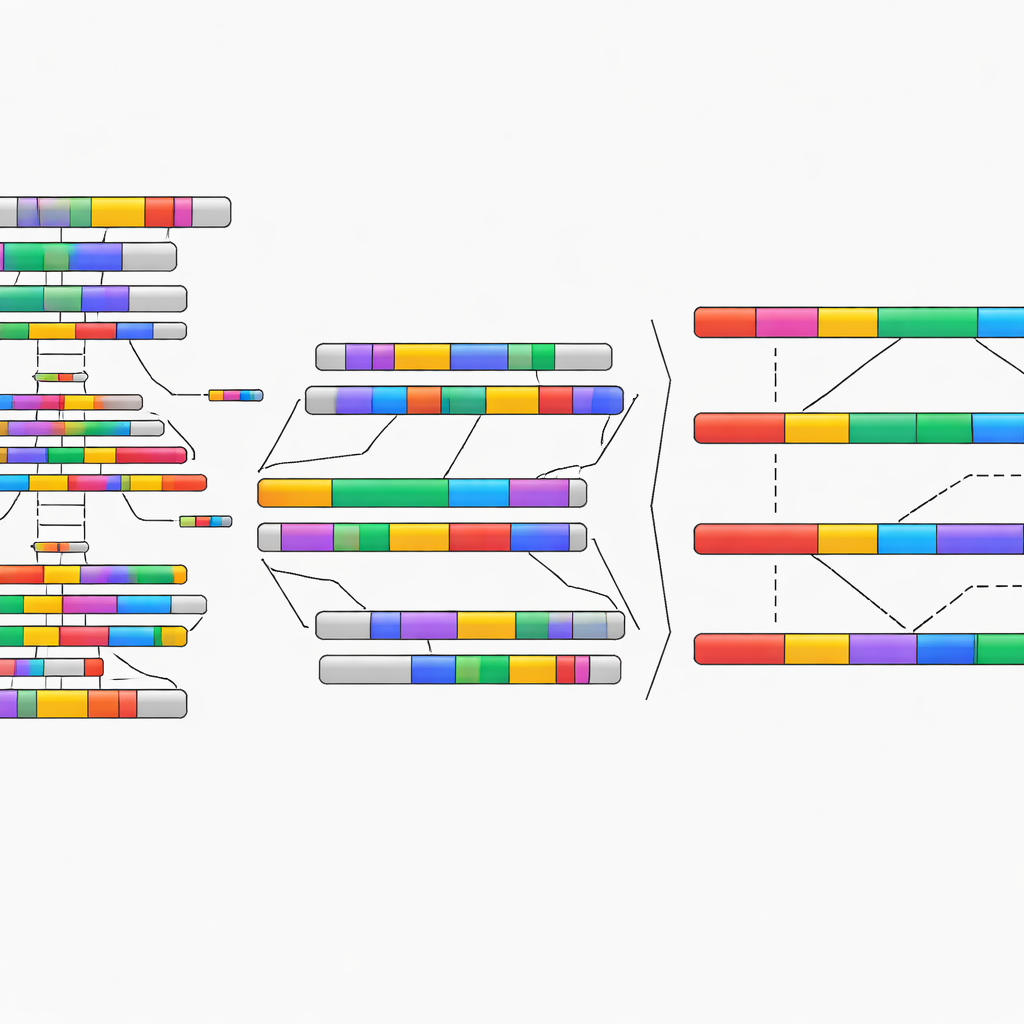
Usar software inteligente para separar copias de genes enredadas
Para abordar los genes que seguían siendo confusos incluso con lecturas largas, los autores recurrieron a una herramienta especializada de faseo llamada Paraphase. En lugar de analizar cada lectura aisladamente, este software agrupa lecturas en “haplotipos” distintos—versiones coherentes de cada copia de un gen—vuelve a alinearlas a un patrón común y rastrea cómo viajan las variaciones de secuencia juntas. Aplicado a 79 grupos de genes altamente similares que se predijo u observaron como difíciles, Paraphase pudo reconstruir haplotipos casi completos y limpios para más de tres cuartas partes de ellos. En ejemplos detallados, como un par de genes implicado en la audición, lecturas que antes estaban mezcladas entre copias pudieron asignarse claramente a pistas separadas, ilustrando cómo la visión algorítmica puede superar los límites de la simple longitud de lectura.
Qué significa esto para el diagnóstico genético futuro
Para los no especialistas, el mensaje principal es que las lecturas de ADN más largas ya marcan una gran diferencia para genes que tienen gemelos confusos, y claramente superan a las pruebas tradicionales de lectura corta en muchas regiones de importancia médica. Sin embargo, incluso las mejores tecnologías actuales de lectura larga no pueden resolver completamente todas las familias de genes enredadas, porque algunos tramos del genoma son simplemente demasiado repetitivos. Este estudio muestra que combinar tres elementos—predicciones informáticas cuidadosas de dónde es probable que haya problemas, datos clínicos reales de lectura larga y software de faseo dedicado—proporciona una hoja de ruta práctica para identificar qué genes se pueden confiar, cuáles requieren precaución adicional y dónde se necesitan con mayor urgencia nuevos métodos. En la genómica clínica, ese tipo de delimitación clara es esencial para convertir la mejora continua en secuenciación en diagnósticos realmente fiables.
Cita: Kim, S.K., Jang, J., Kim, Y. et al. Integrative analysis of in silico predictions and clinical evidence to delineate the capability of HiFi long-read sequencing in paralogous genes. npj Genom. Med. 11, 21 (2026). https://doi.org/10.1038/s41525-026-00555-2
Palabras clave: secuenciación de lectura larga, genes paralogados, genómica clínica, mapeabilidad del genoma, faseo de haplotipos