Clear Sky Science · en
Reduced ULK1 links impaired autophagy and mitophagy to Alzheimer’s disease pathology
Why Brain Recycling Matters as We Age
Alzheimer’s disease robs people of memory and independence, yet the root causes of this slow decline are still being uncovered. This study explores a natural cellular cleaning system that helps brain cells stay healthy. The focus is on a protein called ULK1, which acts like an on switch for the cell’s recycling machinery. By tracking ULK1 in people and in multiple laboratory models, the researchers ask a simple question with big implications: does a fading recycling switch help drive Alzheimer’s disease, and could turning it back up protect the brain?
A Cleanup Switch That Fades with Age
To understand how ULK1 behaves in real people, the team measured it in blood and spinal fluid samples from older adults with normal cognition and from patients with Alzheimer’s at different stages. They found that ULK1 levels dropped over four years even in cognitively healthy volunteers, and were generally lower in people with Alzheimer’s. Brain tissue from donors showed a similar pattern: in key memory regions, neurons from Alzheimer’s brains had less ULK1 than those from age-matched controls. People whose spinal fluid contained more ULK1 at the start of the study tended to show slower worsening of dementia scores over time, hinting that this protein helps buffer the brain against decline. 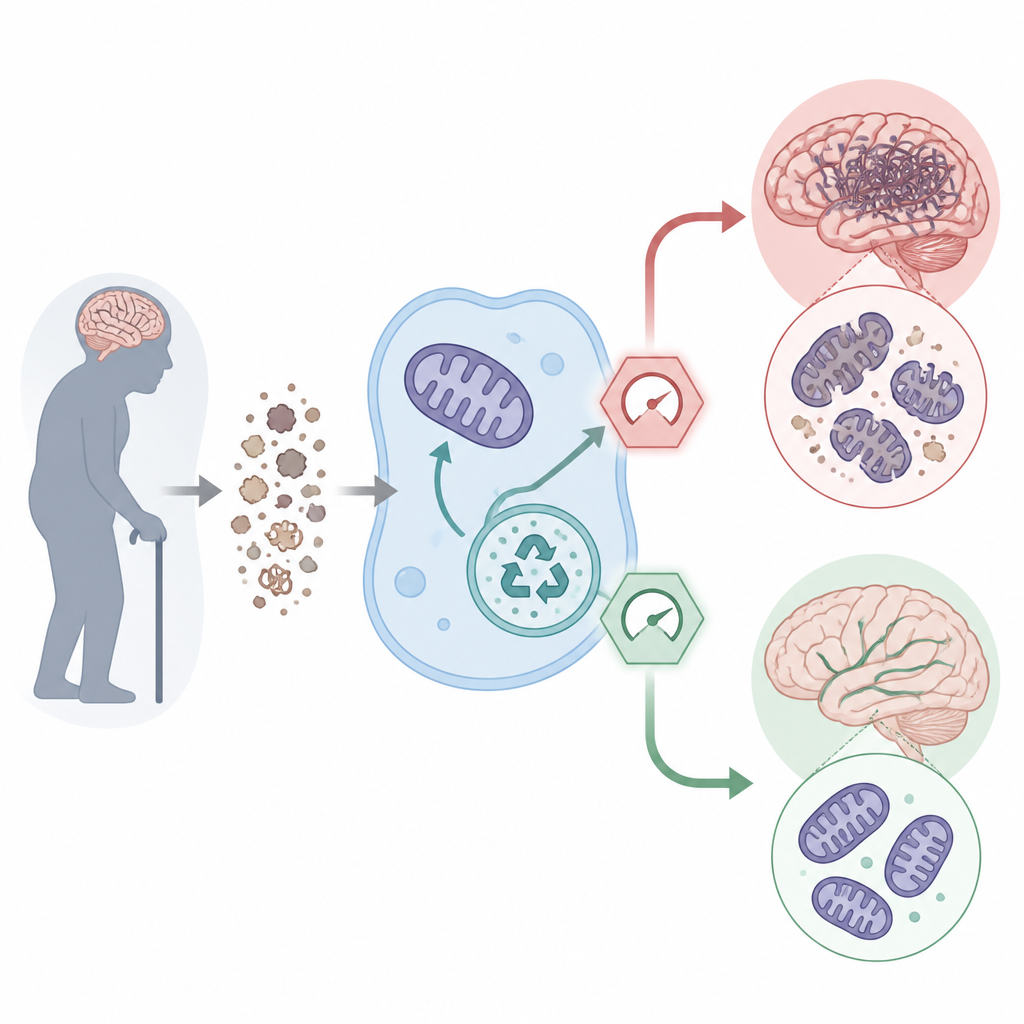
How Weak Recycling Harms Brain Cells
Inside cells, ULK1 starts two related recycling jobs. One clears out damaged proteins and cell parts; the other, called mitophagy, specifically targets worn-out mitochondria, the tiny power plants that fuel brain activity. In Alzheimer’s, sticky protein clumps made of amyloid and tau build up and mitochondria become faulty, creating a vicious cycle of damage. The researchers showed that when ULK1 levels fall, these cleanup systems flag, damaged mitochondria accumulate, and classic Alzheimer’s features such as amyloid plaques and tau tangles worsen. In mice engineered to develop Alzheimer’s-like changes, these defects were tracked at the molecular level and matched to poorer memory performance in maze tests.
Boosting the Switch Protects Memory
Next, the team asked what happens if ULK1 is turned up rather than down. They created mice that make extra ULK1 throughout the body and crossed them with Alzheimer’s strains that develop amyloid plaques or tau tangles. Extra ULK1 did not noticeably change body weight, movement, or basic metabolism, but it had strong effects in the brain. Neurons became more resilient to chemical stress and to toxic amyloid fragments in cell culture. In living mice, ULK1 overexpression improved performance in several memory tasks, reduced the number and size of amyloid plaques, and preserved the density of dendritic spines, the tiny contact points between nerve cells. Microglia, the brain’s clean-up cells, clustered more efficiently around plaques and engulfed more amyloid, while signs of harmful astrocyte activation were reduced. 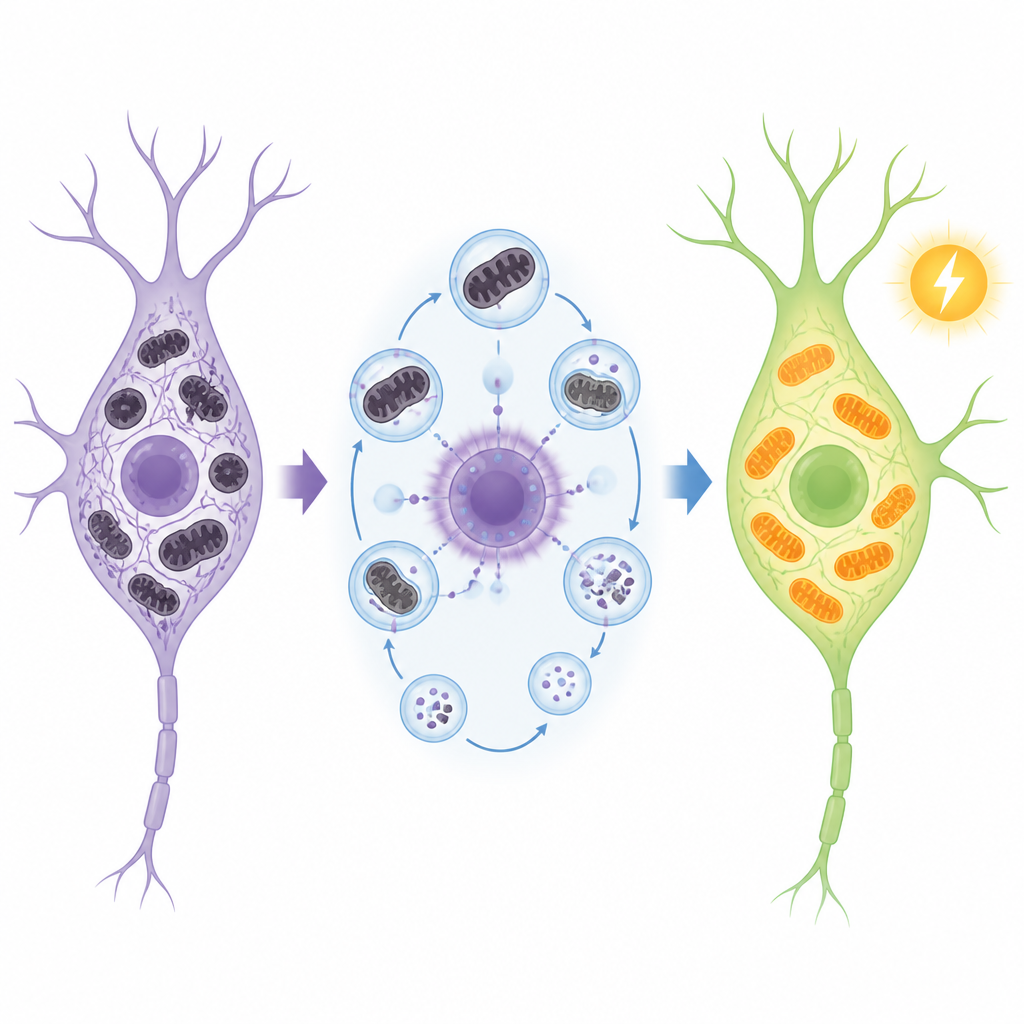
Inside the Mechanism: Power, Fuel, and Tangled Tau
Diving deeper, the researchers found that extra ULK1 restored many genes and pathways related to mitochondrial function and energy production. Electron microscopy revealed fewer damaged mitochondria and more active mitophagy in vulnerable brain regions, accompanied by higher brain ATP levels. ULK1 also influenced tau, the protein that forms internal tangles in Alzheimer’s. In tau-mutant mice, raising ULK1 cut down key tau modifications linked to disease and sharply lowered one particular chemical tag, acetylation at a site called Lys174. This change was tied to higher levels of the cell fuel NAD+ and activation of SIRT1, a protein that removes acetyl groups. In cells, blocking SIRT1 reversed ULK1’s ability to reduce this acetylated tau and to limit tau aggregation, underscoring a chain in which ULK1 boosts recycling, which raises NAD+, which in turn activates SIRT1 to keep tau from becoming dangerously sticky.
Testing Drug-Like Activators Across Species
Because genetic changes are not practical therapies for people, the authors tested small molecules that nudge ULK1 activity up or down. In human cell models where tau clumps can be triggered by adding tau “seeds,” an ULK1 activator called Rac-BL-918 reduced the formation of new tau aggregates and sped removal of existing ones, especially when it strongly stimulated mitophagy. Silencing ULK1, but not its close relative ULK2, erased this benefit, showing that the effect was specific. In the tiny worm Caenorhabditis elegans engineered to express human tau, turning down the ULK1 equivalent or treating with ULK1 inhibitors worsened a simple odor-based memory test. Turning it up genetically or with Rac-BL-918 improved memory, but only if worm versions of key mitophagy proteins were intact. Together, these findings reveal a conserved role for ULK1-driven mitochondrial cleanup in protecting memory across species.
What This Means for Future Alzheimer’s Care
Overall, this work ties a gradual drop in a single cellular switch, ULK1, to failing cleanup systems, rising protein clutter, and memory loss during aging and Alzheimer’s disease. In people, lower ULK1 accompanies disease and may help flag faster progression. In laboratory models, boosting ULK1 strengthens the brain’s own recycling pathways, clears damaged mitochondria and toxic proteins, and improves learning and memory without obvious side effects in midlife animals. While much remains to be tested in humans, ULK1 now stands out as both a candidate biomarker and a potential therapeutic target, pointing to brain recycling and mitochondrial health as central levers in delaying or reducing Alzheimer’s damage.
Citation: Pan, JP., Wang, PJ., Zhang, J. et al. Reduced ULK1 links impaired autophagy and mitophagy to Alzheimer’s disease pathology. Nat Aging 6, 1079–1102 (2026). https://doi.org/10.1038/s43587-026-01108-z
Keywords: Alzheimer’s disease, autophagy, mitophagy, ULK1, tau pathology