Clear Sky Science · en
An acetylation-dependent switch underlies host disease tolerance during streptococcal infection
How Some Infections Hurt Tissues While Others Don’t
Strep throat and severe “flesh-eating” infections are caused by the same bacterium, Streptococcus pyogenes, yet the amount of tissue damage can differ dramatically from case to case. This study asks a deceptively simple question with big implications: why does the same germ sometimes cause overwhelming destruction, and other times remain contained so the body can heal? The researchers show that a tiny biochemical switch, based on how cells use a common building block called acetyl groups, helps decide whether infection spirals into damage or settles into a more tolerable, controlled state.
Two Ways the Body Survives Infection
When we think about fighting germs, we usually picture the immune system killing invading microbes. But survival has a second dimension called disease tolerance: the body’s ability to limit collateral damage so organs keep working even while germs are present. In mouse skin infected with Streptococcus pyogenes, the authors compare normal bacteria with a mutant lacking a key enzyme, pyruvate dehydrogenase (PDH), that reshapes bacterial metabolism. Normal bacteria trigger large, spreading ulcers and dead tissue, while the PDH‑deficient strain produces smaller, well-contained lesions that heal faster. Importantly, both infections initially carry similar numbers of bacteria, showing that the better outcome is not due to stronger killing, but to a host response that is less destructive yet still effective.
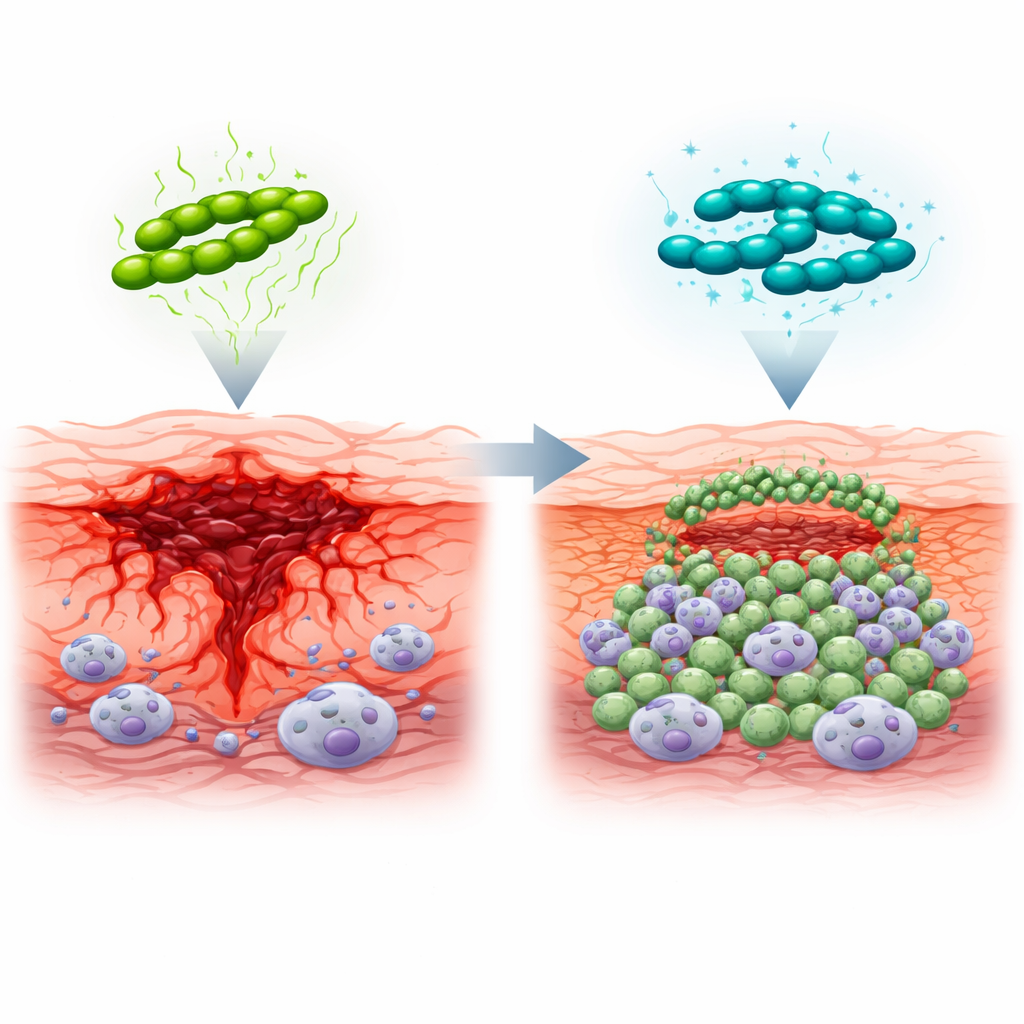
How Bacterial Metabolism Rewires Immune Cells
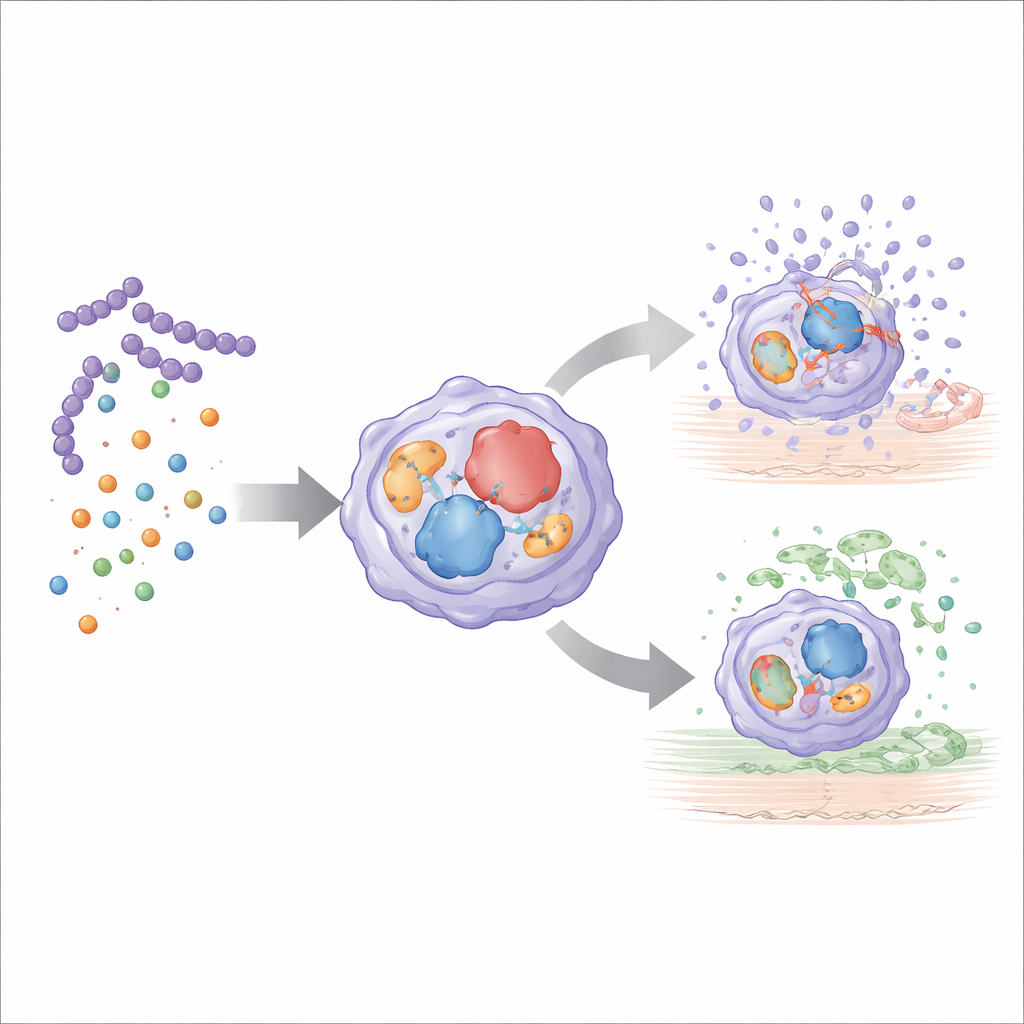
PDH allows the bacteria to convert nutrients into short-chain fatty acids, such as acetate and formate, which spill into surrounding tissues. These small molecules feed into host metabolism, boosting levels of acetyl‑CoA, a central fuel and a key ingredient for protein acetylation inside immune cells. By reanalyzing single-cell RNA sequencing data from infected skin, the team shows that when PDH is missing and these fatty acids are scarce, immune cells shift their energy use: glycolysis (sugar burning) becomes more active, while acetyl‑CoA–linked pathways are dampened. Microscopy reveals that under these conditions, bacteria are trapped alive inside intact phagocytes rather than bursting them, and the overall pattern of signaling between immune cells becomes more coordinated and less self‑amplifying.
From Inflammation to Protection
The metabolic shift in PDH‑deficient infections goes hand in hand with a deep reprogramming of immune cell behavior. Neutrophils and macrophages, the main early responders, expand in number but turn on genes that help manage stress, handle iron safely, and control reactive oxygen species, while dialing down genes that normally chew up tissue and drive runaway inflammation. A broader “disease tolerance” program switches on, including responses to low oxygen, antioxidant defenses, controlled forms of cell death, and pathways involved in wound repair and new blood vessel growth. Rather than simply damping immunity, the response becomes more precise: key inflammatory messengers are still produced to recruit help, but in a balanced way that avoids burning the surrounding tissue.
An Acetylation Switch at the Heart of the Response
Because acetyl‑CoA also supplies acetyl groups for turning proteins on or off, the authors suspected that changes in acetylation link bacterial metabolism to gene activity in immune cells. They revisited bulk RNA sequencing data from infected macrophages treated with an inhibitor of histone deacetylases, enzymes that remove acetyl groups. Blocking these enzymes with the drug Trichostatin A disrupted the protective gene program seen in PDH‑deficient infection and suppressed production of IL‑10, a key anti‑inflammatory and tissue‑repairing signal. At the same time, markers of protein acetylation on histones did not shift dramatically in vivo, pointing instead to acetylation of non‑histone regulators—signaling proteins that act like master dimmer switches for entire networks of genes.
Why This Matters for Treating Dangerous Infections
Altogether, the work outlines a chain of cause and effect: bacterial PDH activity shapes the mix of small fatty acids released during infection, which in turn tunes host acetyl‑CoA levels, protein acetylation, and the behavior of frontline immune cells. When this chain favors a “high‑acetylation” state, inflammation becomes more damaging; when it is restrained, immune cells still contain bacteria but protect tissue through disease tolerance programs. By revealing acetylation as a central control knob that couples microbial metabolism to host defenses, the study suggests new treatment strategies: instead of just killing bacteria, we might design drugs that gently steer this metabolic‑epigenetic switch to keep tissues safe while the immune system does its job.
Citation: Paudel, S.K., Gannavaram, S., Caparon, M.G. et al. An acetylation-dependent switch underlies host disease tolerance during streptococcal infection. Sci Rep 16, 11947 (2026). https://doi.org/10.1038/s41598-026-42565-1
Keywords: disease tolerance, Streptococcus pyogenes, immunometabolism, acetylation, host–pathogen interactions