Clear Sky Science · en
Targeted deletion of c-kit in TECs attenuates UUO-induced renal fibrosis through NF-κB pathway inhibition
Why this study matters for kidney health
Chronic kidney disease affects hundreds of millions of people worldwide, and once scar tissue builds up in the kidney, the damage is usually permanent. This study asks a simple but important question: can we slow or soften that scarring by switching off one specific signal in the cells that line the kidney’s tiny tubes? By tracing how this signal drives inflammation and scar formation in mice, the researchers point to a potential new way to protect kidney function before it is too late.
A closer look at scarring inside the kidney
In most long‑term kidney disease, the key problem is not just damage to the blood‑filtering units, but also a gradual thickening and stiffening of the tissue between them, known as fibrosis. As this tissue fills with collagen and other fibers, the kidney shrinks, hardens, and loses its ability to clean the blood. The cells that line the kidney tubules sit right in the middle of this process: they sense injury, send out distress signals, and can even change shape toward a more fibrous, scar‑producing state. Understanding which signals push these cells toward scarring is crucial for finding new treatments.
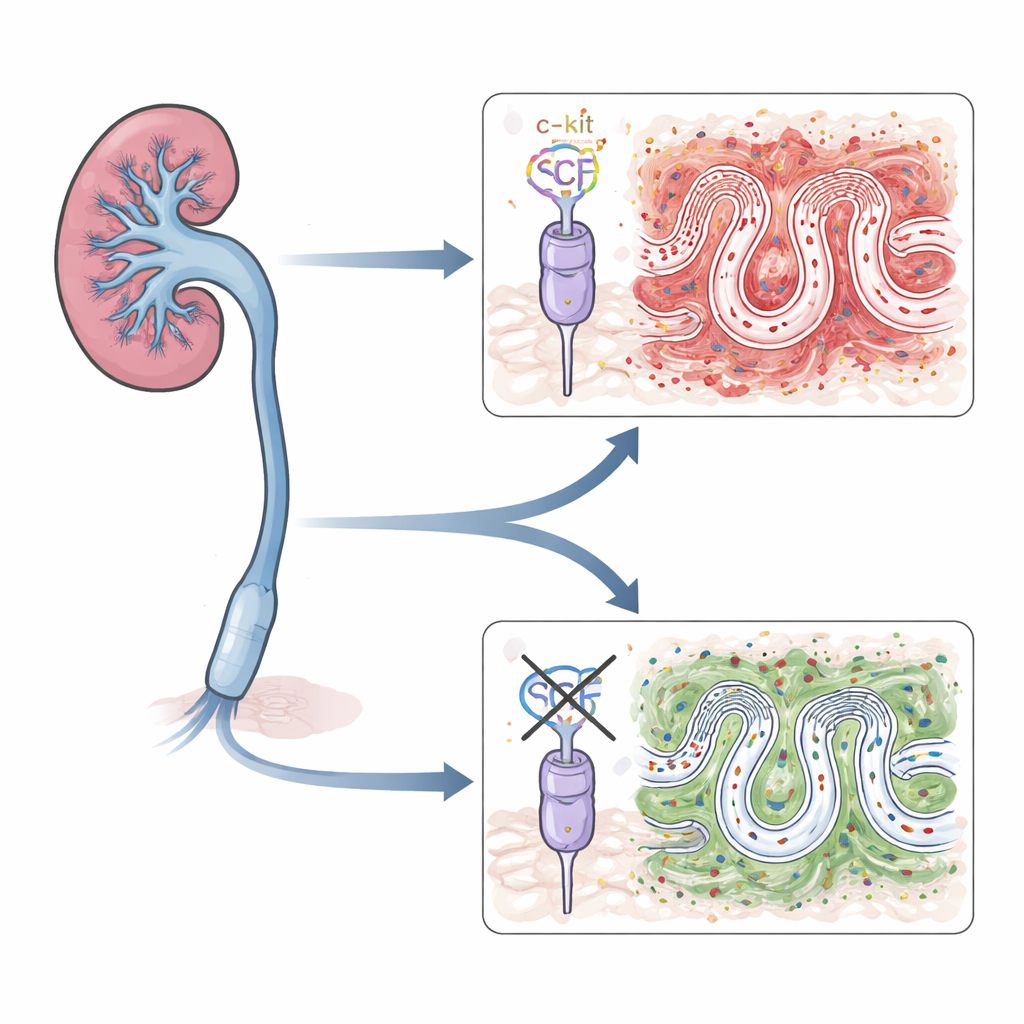
A signal pair under suspicion
The team focused on a well‑known pair of molecules: stem cell factor (SCF) and its receptor, c‑kit. Together they help control the growth and movement of several cell types in the body and have been linked to scarring in the lung, liver, and other organs. In diseased kidneys, both SCF and c‑kit are found at higher levels, and earlier work suggested they might worsen fibrosis mainly by activating immune cells called mast cells. However, tubular epithelial cells themselves also carry this receptor. The authors set out to test whether SCF acting directly on these tubule cells is enough to drive kidney scarring.
Switching off c‑kit in tubule cells
To tease apart this question, the researchers engineered mice in which c‑kit is deleted only in kidney tubular cells, leaving the rest of the body’s c‑kit signals intact. They then used a standard procedure called unilateral ureteral obstruction, in which one urine‑carrying tube from the kidney is tied off. This reliably causes pressure, inflammation, and fibrosis in that kidney over days to weeks. In normal mice, the blocked kidneys showed rising levels of SCF, c‑kit, and several proteins that mark scar‑forming cells, along with heavy collagen deposits. In contrast, mice lacking c‑kit only in their tubule cells had milder tissue damage and significantly less collagen buildup, even though the obstruction was the same.
Connecting the signal to inflammation
The study also probed how SCF and c‑kit feed into a master switch for inflammation inside cells, known as the NF‑κB pathway. By analyzing gene‑activity datasets and tracking key proteins in this pathway, the authors found that kidneys with high c‑kit activity also showed stronger NF‑κB activation and higher levels of inflammatory messengers such as IL‑6 and IL‑1β. In the engineered mice lacking tubular c‑kit, activation of this pathway and the associated inflammatory signals were clearly blunted after obstruction. This suggested that SCF and c‑kit in tubule cells amplify both inflammation and scarring through NF‑κB.
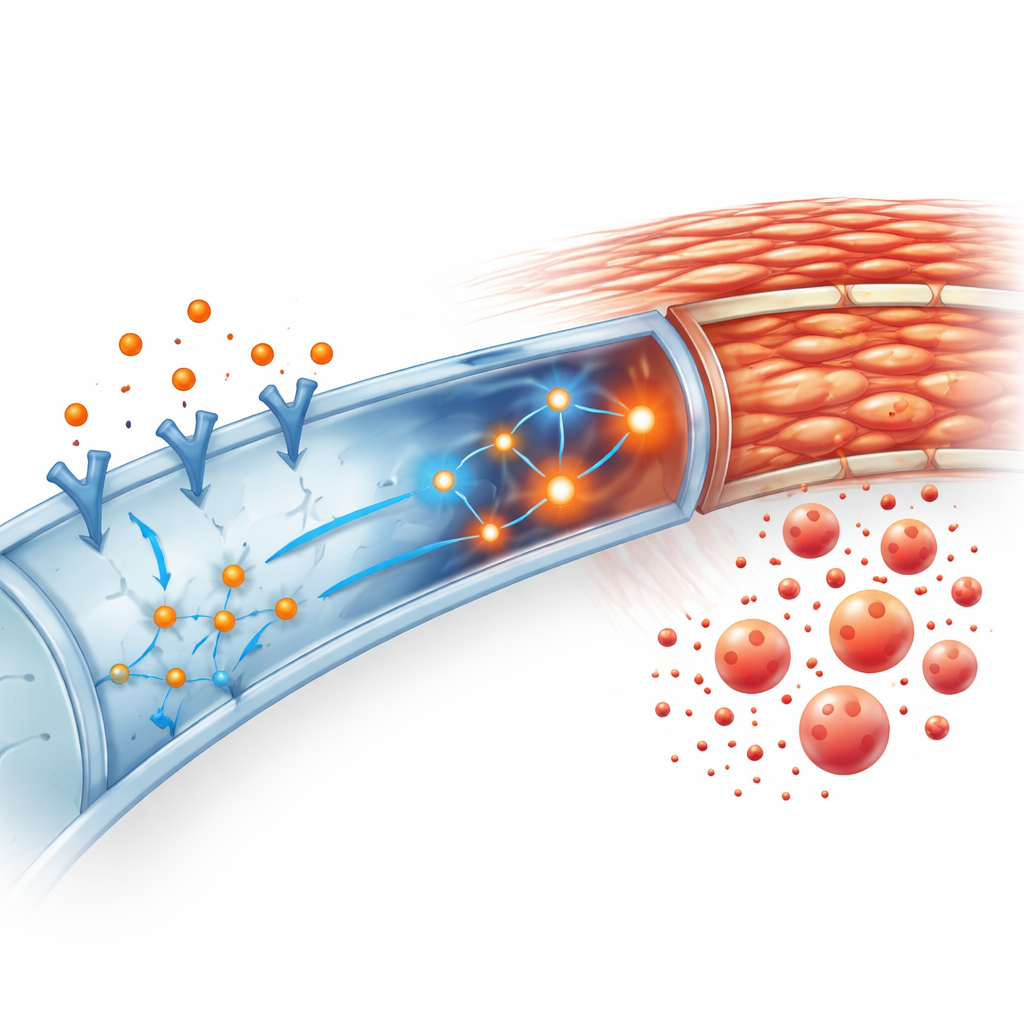
Testing the process in isolated kidney cells
To confirm that this was a direct effect on tubule cells, the team grew primary tubular epithelial cells from normal and genetically altered mice in the lab. When they added SCF to normal cells, the cells adopted a more elongated, scar‑like appearance and sharply increased their production of fibrosis‑related proteins. These changes came with a surge in NF‑κB activity. Cells that lacked c‑kit did not show the same fibrotic shift after SCF. Moreover, when the researchers blocked NF‑κB with a specific drug, SCF could no longer push normal tubule cells toward a fibrotic state. Together, these experiments support a chain of events: SCF activates c‑kit on tubule cells, which in turn activates NF‑κB, driving inflammation and collagen production.
What this could mean for future treatments
In everyday terms, this work identifies a kind of “ignition switch” for scarring inside the kidney tubules. By turning off c‑kit only in these cells, the researchers reduced early loss of kidney function, cut down on inflammatory signals, and limited the buildup of scar tissue in a well‑established mouse model. While more studies are needed to confirm how this pathway behaves in human kidneys and to test existing c‑kit–blocking drugs for safety in this context, the findings suggest that carefully targeting the SCF/c‑kit–NF‑κB axis in tubule cells could help slow the march from kidney injury to irreversible chronic kidney disease.
Citation: Xing, Z., Wang, H., Zhou, Y. et al. Targeted deletion of c-kit in TECs attenuates UUO-induced renal fibrosis through NF-κB pathway inhibition. Sci Rep 16, 13227 (2026). https://doi.org/10.1038/s41598-026-42540-w
Keywords: chronic kidney disease, renal fibrosis, tubular epithelial cells, c-kit signaling, NF-kappaB