Clear Sky Science · en
Mutation type, tyrosine kinase function in normal cell and tyrosine kinase inhibitor activity in cancers
Why some smart cancer drugs work better than others
In recent years, cancer medicine has been transformed by pills that switch off overactive growth signals inside tumor cells. These drugs, called tyrosine kinase inhibitors, can lead to dramatic and long-lasting remissions—but not for every patient, even when tumors carry seemingly similar gene changes. This study asks a deceptively simple question with big consequences for patients and drug development: can we predict which cancers will truly depend on a given growth signal, and therefore respond well to a matching targeted drug?
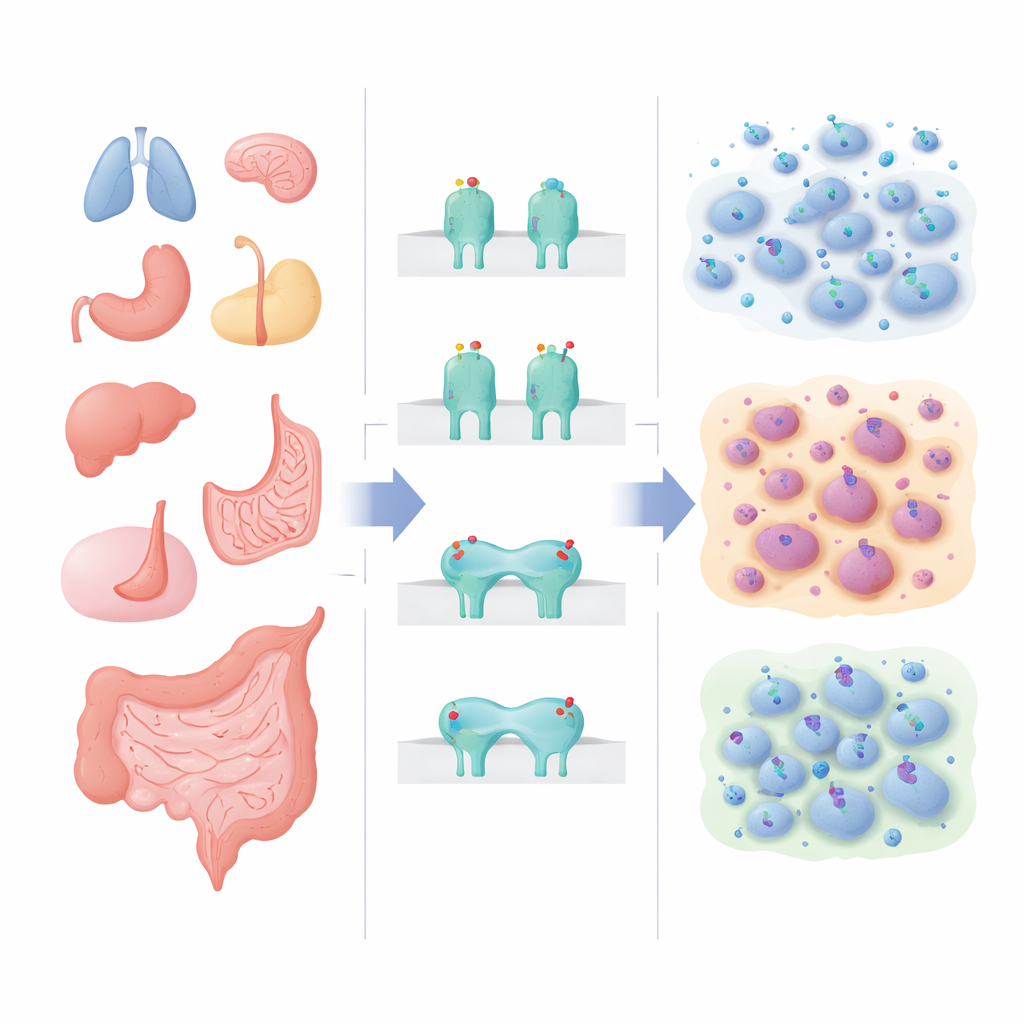
Targeted pills and their mixed track record
Tyrosine kinases are molecular on-off switches that help cells respond to their environment. When the genes encoding these switches are altered, they can drive cancer growth. Drugs designed to block faulty kinases—such as those targeting EGFR, KIT, ALK, or TRK—have revolutionized care for several cancers. Yet the same type of genetic alteration in the same kinase can lead to very different drug responses depending on the cancer. For example, patients with gastrointestinal stromal tumors carrying certain KIT mutations can enjoy years of disease control on imatinib, while people with melanomas bearing similar KIT mutations often see only brief benefit. Such inconsistencies have fueled interest in “tissue-agnostic” therapies that work across many tumor types, but also highlighted how incomplete our current rules are.
Four data lenses on the same problem
The researchers assembled a large, integrative picture using four kinds of public data. First, they reviewed 35 clinical trials of the best available kinase inhibitors used alone in 43 specific combinations of cancer type, tyrosine kinase, and mutation class. They focused on median progression-free survival—the time before tumors began to grow again—along with response rates. Second, they examined how often 16 key receptor tyrosine kinases were altered by three mechanisms—single-letter “missense” mutations, gene amplifications, or gene fusions—across 17 common and rare solid cancers in the MSK-IMPACT sequencing cohort. Third, they looked up how strongly each kinase is normally expressed in healthy tissues using the GTEx project. Finally, they scored how essential each kinase is for normal cell development and maintenance, drawing on human genetic syndromes and mouse knockout or knock-in experiments compiled in the OMIM database and the scientific literature.
Not all gene changes are equal
By comparing these layers, several clear patterns emerged. Across all cancers and kinases, gene fusions—where part of a kinase gene becomes joined to another gene—were associated with the longest benefit from kinase inhibitors. Missense mutations produced intermediate results, while simple gene amplifications rarely yielded strong responses to single-agent drugs. How common a given alteration was in a cancer type did not reliably predict drug success, nor did how much of the kinase’s messenger RNA was present in the corresponding healthy tissue. The mere presence or frequency of a mutation, it seems, is not enough to guarantee that a tumor is truly addicted to that signal or that blocking it will matter clinically.
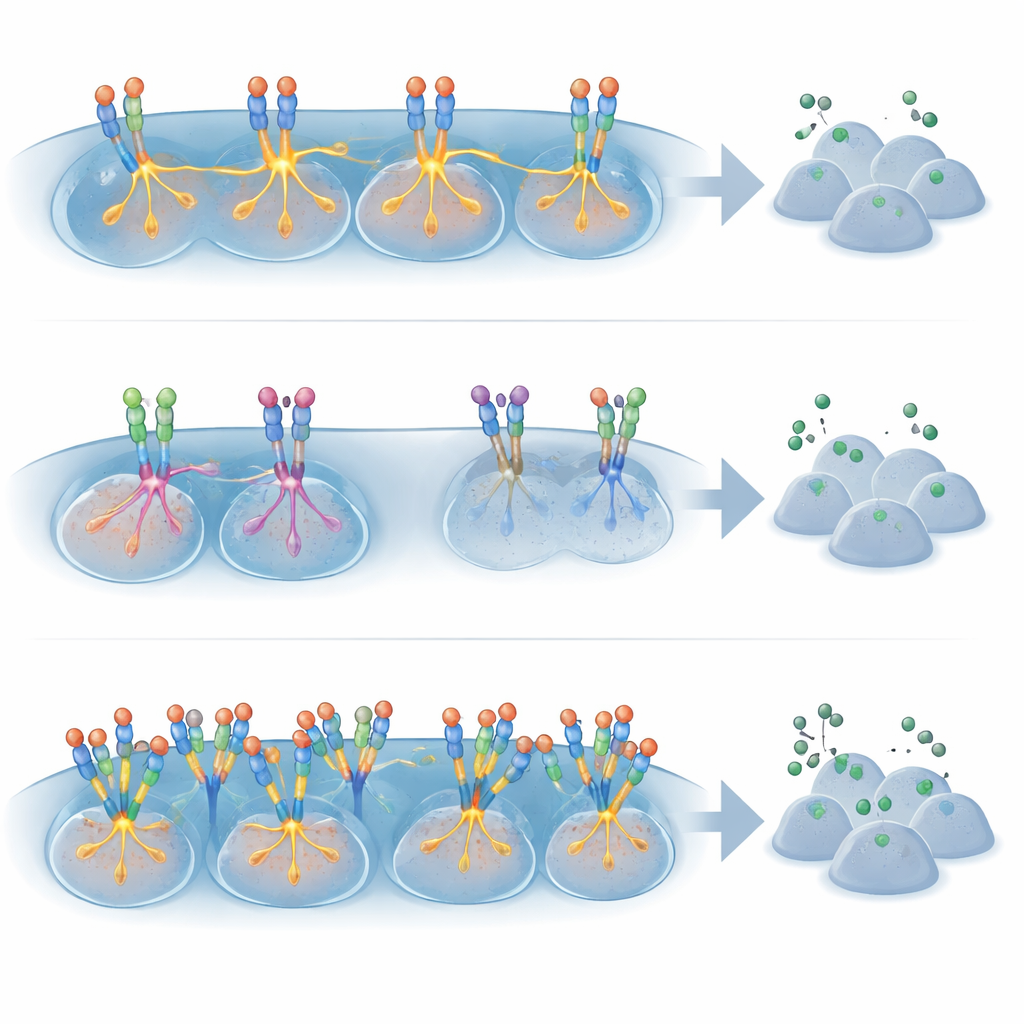
Normal cell “importance” as a hidden guide
The most striking insight came when the team considered how important a kinase is for the normal cell type from which the cancer arises. They classified each kinase–tissue pair as having a strong, probable, absent, or unclear role based on genetic evidence. For tumors driven by missense mutations, kinase inhibitors worked best when the same kinase plays a major role in the matching healthy tissue—for instance, KIT in the specialized gut cells that give rise to gastrointestinal stromal tumors, or EGFR in lung epithelial cells. In these settings, the mutation appears to hijack an already central control system, making the cancer highly dependent on it. In contrast, for gene fusions, effective drug responses appeared largely independent of any normal role: fusion-driven tumors responded well even when the kinase is barely used in the original tissue. Amplifications again stood out as poor predictors of benefit, regardless of normal function.
What this means for patients and future drugs
For people with cancer, these findings help explain why some targeted pills can be life-changing while others offer only modest delays. When a single-letter mutation affects a kinase that is already crucial for the healthy version of that cell type, the cancer is more likely to be vulnerable to a matching inhibitor. When a kinase is overexpressed but not central to normal cell function—or simply amplified in extra copies—the tumor may rely on many other pathways and shrug off the drug. Fusion events are a special category: they often turn the kinase into a powerful, stand-alone engine of growth, so blocking them tends to work across multiple tissues. Together, this work suggests a practical roadmap for precision oncology: prioritize drug development and clinical trials for rare fusion-driven cancers, and for missense mutations in kinases with proven importance in the originating tissue, while treating amplifications more cautiously as drug targets.
Citation: Vanacker, H., Cassier, P.A., Dufresne, A. et al. Mutation type, tyrosine kinase function in normal cell and tyrosine kinase inhibitor activity in cancers. Sci Rep 16, 11968 (2026). https://doi.org/10.1038/s41598-026-41710-0
Keywords: tyrosine kinase inhibitors, precision oncology, oncogene addiction, fusion-driven cancer, targeted cancer therapy