Clear Sky Science · en
Crystal structure of human INPP5K with an allosteric inhibitor reveals the structural basis for species specific potency
Why controlling this muscle enzyme matters
Our muscles rely on a finely tuned chemical system to respond to insulin, take up sugar from the blood, and grow or repair after exercise. One of the key regulators in this system is an enzyme called INPP5K, which acts as a brake on signals that promote glucose uptake and muscle formation. Scientists have been searching for small molecules that can dial this brake up or down, both to better understand muscle biology and to explore future treatments for conditions such as insulin resistance, muscle wasting, and some brain tumors. This study uncovers, in atomic detail, how a new drug-like compound latches onto human INPP5K in an unexpected way—and why it works in some animals but not others.
A molecular brake on insulin signals
INPP5K is found in high amounts in skeletal muscle, as well as in the brain, heart, and kidney. It acts on a special class of fatty molecules in cell membranes that carry signals triggered by hormones like insulin and growth factors. When these membrane signals are strong, muscle cells pull more glucose out of the blood and are pushed toward growth and differentiation. INPP5K trims these signaling molecules, weakening the message and keeping insulin responses in check. Mice that lack one working copy of the INPP5K gene become unusually sensitive to insulin and develop larger muscles, and patient studies have linked INPP5K mutations to certain congenital muscular dystrophies. These clues have made the enzyme an attractive target for chemical probes and, potentially, drugs.
Finding a subtle off-switch
Using high-throughput screening, the researchers previously discovered a small molecule, called CPD-1, that blocks human INPP5K activity at very low concentrations. Intriguingly, CPD-1 does not compete with the natural signaling lipid for the usual active site; instead, it acts as a noncompetitive inhibitor, suggesting it works through a subtler, long-range effect on the protein’s shape. Even more surprising, the compound is strongly effective against human INPP5K but barely affects the versions found in mice and rats. To unravel both puzzles—how CPD-1 inhibits the enzyme, and why it is so selective—the team turned to X‑ray crystallography, which can reveal the three-dimensional arrangement of atoms in protein–drug complexes.
Revealing a hidden pocket
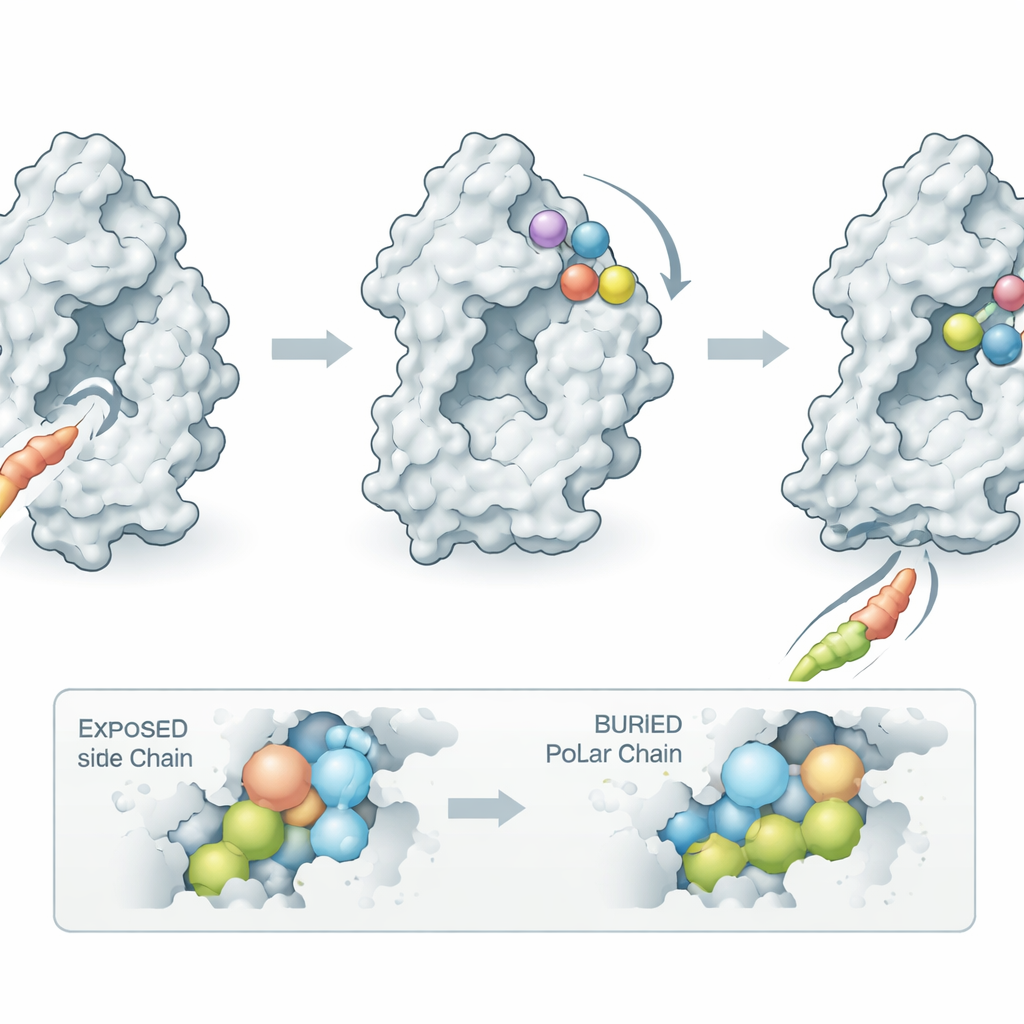
To obtain crystals suitable for structural analysis, the researchers engineered a slightly trimmed version of INPP5K, shortening a floppy surface loop that made the protein difficult to crystallize while keeping its activity and drug sensitivity intact. They then solved the structure of this human enzyme fragment bound to CPD-1 at 1.9‑angstrom resolution, fine enough to see individual side chains and the precise pose of the compound. The structure showed that CPD-1 nestles into a previously unknown pocket on the side of a long helix rather than in the main catalytic groove. Binding there acts like a wedge: it forces that helix to tilt outward by about 22 degrees, which in turn tugs on nearby loops that normally cradle the signaling lipid. In the drug-bound state, these loops are displaced, and the substrate can no longer dock properly, explaining why the inhibitor shuts the enzyme down without sitting in the active site itself.
Why humans and hamsters respond, but mice do not
The structure also clarifies the mystery of species selectivity. The pocket where CPD-1 binds is lined with amino acids that are almost identical in human, mouse, and rat INPP5K, so direct contact differences cannot explain the large gap in potency. Instead, the key lies at the base of the tilting helix, which acts as a hinge. In human INPP5K, this hinge includes a polar residue (glutamine at position 171) that tolerates being exposed to water when the helix swings open to create the pocket. In mice and rats, the same position is occupied by a greasy leucine that prefers to stay buried inside the protein; pulling this hydrophobic side chain into solvent would be energetically costly. As a result, the helix in rodent enzymes is effectively locked in the closed state, and the allosteric pocket that CPD-1 requires never forms efficiently. By comparing sequences from other species, the authors predicted—and confirmed experimentally—that hamster INPP5K, which shares the flexible hinge with humans, is sensitive to CPD-1, whereas guinea pig INPP5K, with extra electrostatic “braces” that hold the helix shut, is only weakly affected.
From structural insight to better models and medicines
By connecting high-resolution structure, biophysics, and enzyme assays, this work shows that CPD-1 disables INPP5K not by plugging its active site but by prying open a distant helix and disturbing the architecture of the substrate-binding region. The discovery of this hidden, druggable pocket explains why the inhibitor is highly selective for INPP5K among related enzymes and why it works in humans and hamsters but not in common laboratory rodents. Practically, the study points to the hamster as a more faithful small-animal model for testing future INPP5K-targeting compounds in vivo. More broadly, the structure offers a template for designing next-generation molecules that fine-tune this muscle- and brain-relevant enzyme, potentially aiding research on insulin sensitivity, muscle degeneration, and invasive cancers where INPP5K activity is implicated.
Citation: Nomura, A., Yamaguchi, K., Kawano, M. et al. Crystal structure of human INPP5K with an allosteric inhibitor reveals the structural basis for species specific potency. Sci Rep 16, 11132 (2026). https://doi.org/10.1038/s41598-026-40748-4
Keywords: INPP5K, allosteric inhibitor, crystal structure, species selectivity, drug design