Clear Sky Science · en
MTCH2 promotes BAX and BAK self-assembly and apoptotic pore growth
Why cell death matters to health
Every day, billions of our cells quietly retire so that tissues stay healthy and damaged cells do not turn cancerous. This controlled self-destruction, called programmed cell death, depends on tiny pores that open in the membranes of mitochondria, the cell’s power plants. The study explored how a little-known helper protein, MTCH2, shapes these pores, influencing not only whether cells die but also how they alert the immune system and respond to infections and cancer drugs.
The cell’s point of no return
When a cell commits to die, a critical step occurs in its mitochondria: the outer membrane becomes perforated, allowing key molecules to spill into the rest of the cell. Two related proteins, BAX and BAK, are central actors in this drama. They cluster on the mitochondrial surface and assemble into large structures that punch holes in the membrane. These openings let out signals that activate destructive enzymes, but they also release fragments of mitochondrial DNA that can rouse immune defenses. Until now, researchers knew little about other cellular components that might control how these pores form and grow.
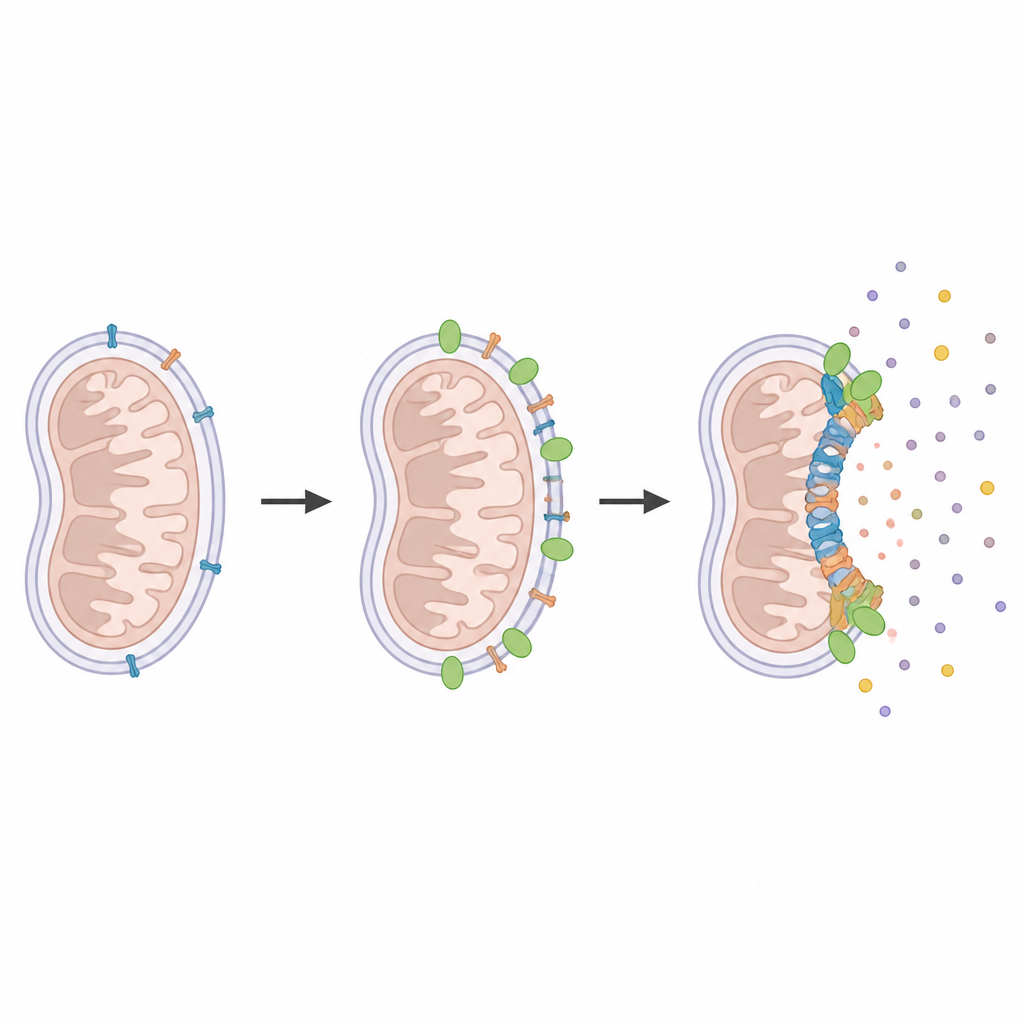
Finding neighbors at the death pore
To identify proteins that gather around BAX and BAK during cell death, the researchers used a clever molecular tagging strategy. They fused an enzyme called APEX2 to BAX, BAK, or another mitochondrial protein and then briefly activated it only in living cells. APEX2 marked any nearby proteins within a few billionths of a meter, which were later fished out and identified by mass spectrometry. Comparing healthy and dying cells revealed a shortlist of proteins that specifically cluster near the forming pores. Among them, MTCH2 stood out as a recurring neighbor of both BAX and BAK under death-inducing conditions.
A helper for pore building
Next, the team asked what happens if cells lack MTCH2. Using advanced microscopy to watch single clusters of BAX and BAK in real time, they observed that mitochondria still lost their electrical charge, but the assembly of large BAX and BAK structures was delayed and less robust. In other words, the point-of-no-return signal appeared on schedule, yet the pores that normally follow grew more slowly and remained smaller. Adding back MTCH2 restored normal pore growth, and supplying cells with a lipid molecule called lysophosphatidic acid could partially compensate when MTCH2 was missing, pointing to a role for membrane fats in the process.
Lipids, DNA escape, and immune alarms
Because MTCH2 has been linked to lipid metabolism, the authors examined the fat composition of mitochondria. They found that cells missing MTCH2 had lower levels of several key phospholipids, including cardiolipin, a molecule known to support pore formation. Treating cells with lysophosphatidic acid boosted specific lipids and rescued BAX and BAK clustering. The team then tracked mitochondrial DNA leaving the organelle and activating an immune-sensing pathway called cGAS–STING. Cells without MTCH2 released less mitochondrial DNA and showed weaker activation of this pathway, as well as altered reshaping of the internal mitochondrial folds that normally accompany DNA escape.
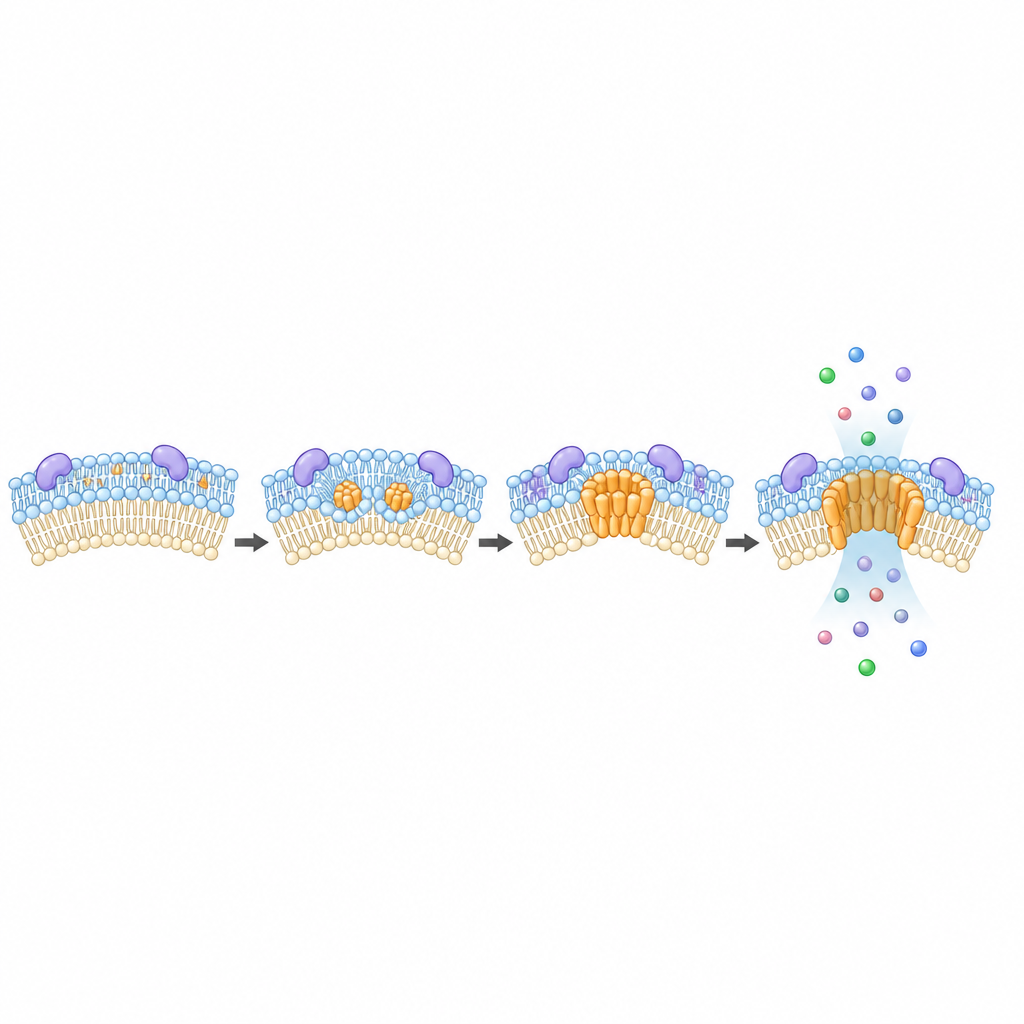
Implications for infection and cancer therapy
The impact of MTCH2 reached beyond laboratory death triggers. When cancer cells were exposed to drugs that only partially engage the cell death machinery, those lacking MTCH2 were more likely to survive and became more resistant to later treatments. In gastric cells infected with the bacterium Helicobacter pylori, which is known to cause subtle mitochondrial damage and DNA injury, MTCH2-deficient cells showed reduced markers of DNA damage compared with normal cells. These findings suggest that MTCH2 helps tune how strongly cells respond to stressful signals that fall short of outright death.
What this means for future treatments
Overall, the work shows that MTCH2 acts as a key organizer of the death pores formed by BAX and BAK, largely by shaping the lipid environment of the mitochondrial membrane. For a lay reader, this means that a single helper protein can influence whether pores are small leaky pinholes or wide gateways that let out powerful signals, including mitochondrial DNA. Because these signals affect inflammation, infection responses, and the survival of cancer cells after therapy, understanding MTCH2 may open paths to medicines that fine-tune cell death rather than simply turning it on or off.
Citation: Flores-Romero, H., Pena-Blanco, A., Aufdermauer, J. et al. MTCH2 promotes BAX and BAK self-assembly and apoptotic pore growth. Nat Struct Mol Biol 33, 824–837 (2026). https://doi.org/10.1038/s41594-026-01805-8
Keywords: apoptosis, mitochondria, BAX BAK, MTCH2, mitochondrial DNA release