Clear Sky Science · en
Stepwise transcription stalling by the anti-cancer drug Actinomycin D and insights into short tandem repeat transcription inhibition
Why stopping harmful genetic messages matters
Our cells constantly copy DNA into RNA, creating the messages that tell the body which proteins to make. But some stretches of DNA made of short, repeated sequences can produce toxic RNA that contributes to more than 60 human diseases, from muscular disorders to neurodegeneration and cancer. This study unpacks, in atomic detail, how an old anti‑cancer drug called actinomycin D can bring this copying process to a halt—especially on these risky repeat regions—offering clues for designing safer, more precise therapies.
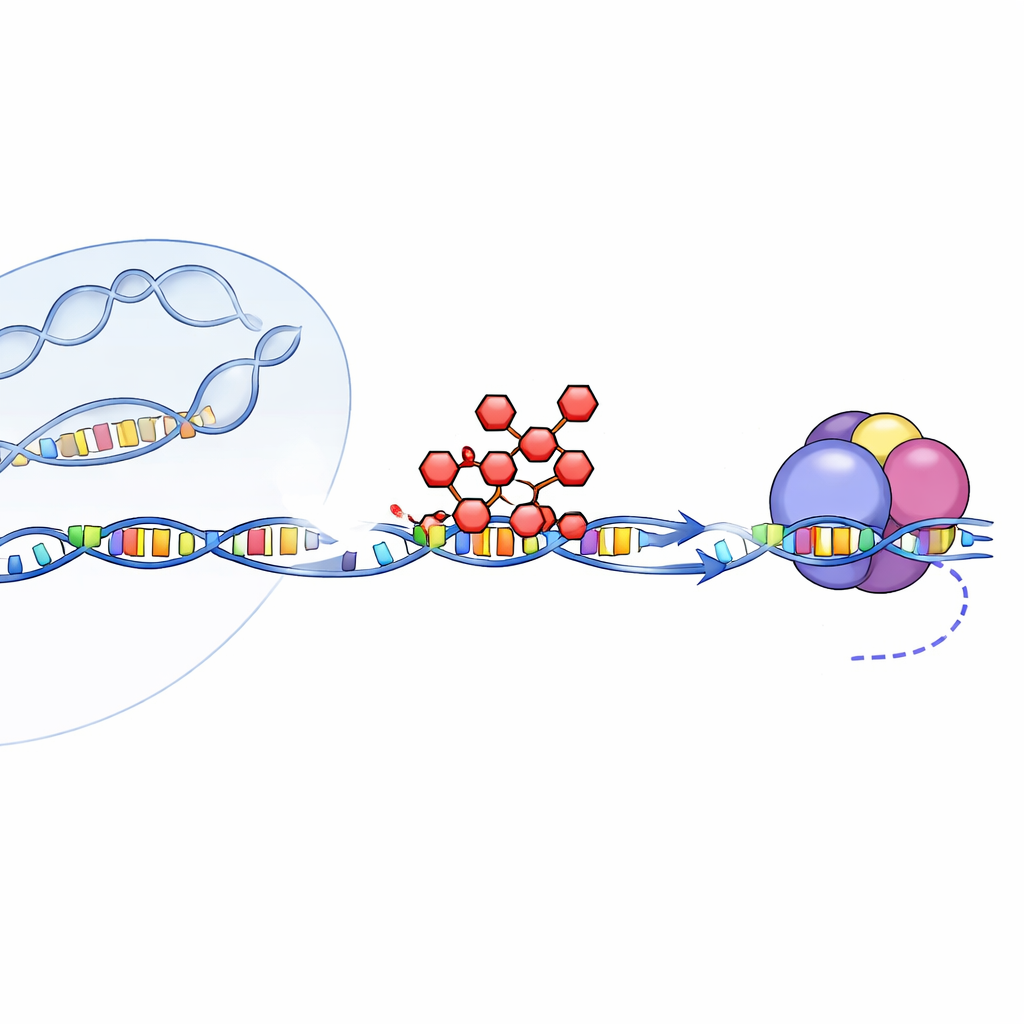
Repeating DNA and toxic cellular clutter
Short tandem repeats are runs of a few DNA letters repeated over and over, and they make up about 6% of our genome. When the cell’s copying machine, RNA polymerase II, reads these regions, the resulting RNA can clump inside the nucleus and trap important RNA‑binding proteins. In diseases like myotonic dystrophy type 1 and Huntington’s disease, expanded repeats such as CTG or CAG produce RNAs and proteins that poison cells. Cancer cells also often crank up transcription, including from repeat‑rich regions, to support uncontrolled growth. Because of this, scientists have long hoped to selectively dial down transcription through these troublemaking stretches.
An old drug with a mysterious braking system
Actinomycin D has been used for decades to treat certain childhood cancers by blocking transcription, but its precise braking mechanism—and its side effects—have been poorly understood. The drug is known to wedge itself between base pairs in DNA, especially at GC‑rich spots, like a shim in the minor groove. Here, the researchers rebuilt simplified yeast and mammalian transcription systems in the lab, using purified RNA polymerase II and synthetic DNA templates with defined actinomycin D binding sites. This setup let them watch, with great control, how the enzyme behaves as it encounters the drug, and then visualize the process at near‑atomic resolution using cryo‑electron microscopy.
Three distinct pause points along the track
The team discovered that RNA polymerase II does not simply crash into actinomycin D once and stop. Instead, it pauses at three distinct positions—called n‑5, n‑2, and n‑1—just upstream of the drug‑bound site. At the earliest “encounter” stage (n‑5), the enzyme has room to add the next RNA building block, but the DNA ahead is already distorted by the drug and starting to interact with a flexible protein segment known as the switch‑1 motif. In the later “engaged” stage (n‑2), the active site is no longer open, and the drug sits tightly sandwiched between the DNA and switch‑1. In the final “stalled” stage (n‑1), actinomycin D also presses against another key structural element, the bridge helix, physically blocking the enzyme’s forward motion and nudging it to slip backward along the DNA. Biochemical tests confirmed that, in the presence of the drug, the enzyme is more prone to backtracking and long‑lived arrest.
How repeats and multiple drug molecules amplify the block
The same stepwise stalling mechanism held true for mammalian (bovine) RNA polymerase II, suggesting that the findings are directly relevant to human cells. The authors then turned to disease‑linked repeats rich in GC sites, including CTG, CAG, CGG, CCUG, and GGGGCC sequences. In controlled reactions, actinomycin D most strongly inhibited transcription of CTG and CAG repeats, matching earlier measurements that showed the drug binds those sequences most tightly. When multiple CTG repeats were present, several actinomycin D molecules could line up on the same stretch of DNA. Structural snapshots with one, two, and three drug molecules bound showed that these neighbors stabilize each other through hydrophobic contacts, increasingly distort the downstream DNA, and progressively weaken its grip on the polymerase. As a result, lower drug doses are needed to stall transcription when GC‑rich repeats cluster together.
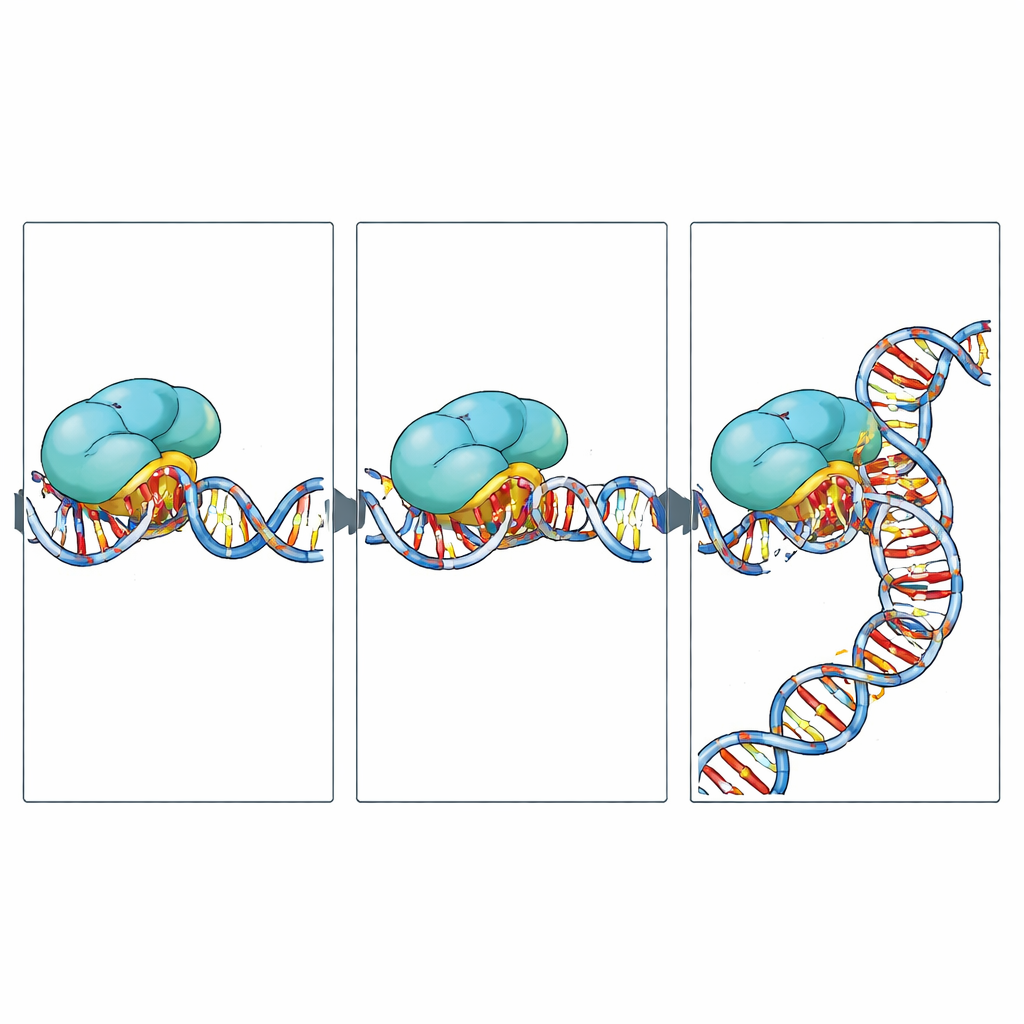
Designing gentler brakes for repeat‑driven diseases
By revealing exactly how actinomycin D wedges into repeat DNA and jams the copying machinery in a series of steps, this work turns a blunt, toxic chemotherapeutic into a blueprint for more refined tools. The structures pinpoint which parts of the drug touch the enzyme and the DNA, and how multiple molecules cooperate on repeated sequences. That information could guide chemists and computational designers to tweak actinomycin D’s peptide rings to strengthen its grip on harmful repeats while easing its impact elsewhere in the genome. In the long run, such tailored molecules might offer new ways to quiet toxic RNAs in neurological disorders and to rein in runaway transcription in cancer, with fewer side effects than the classic drug.
Citation: Zhao, W., Zhu, L., Liu, Y. et al. Stepwise transcription stalling by the anti-cancer drug Actinomycin D and insights into short tandem repeat transcription inhibition. Nat Commun 17, 4005 (2026). https://doi.org/10.1038/s41467-026-70612-y
Keywords: actinomycin D, RNA polymerase II, short tandem repeats, transcription inhibition, repeat expansion diseases