Clear Sky Science · en
Vascular smooth muscle cell state trajectories mediate molecular mechanisms of coronary disease risk
Why the hidden life of artery cells matters
Heart attacks often seem to strike out of the blue, but decades of slow changes in the walls of our arteries set the stage. This study asks a deceptively simple question with big implications: what exactly are the muscle cells in our arteries doing as plaques form and grow, and how do our genes steer those changes toward protection or danger? By following individual cells in mouse arteries over time and tying their behavior to human genetics, the researchers uncover cell "storylines" that help explain inherited risk for coronary artery disease.
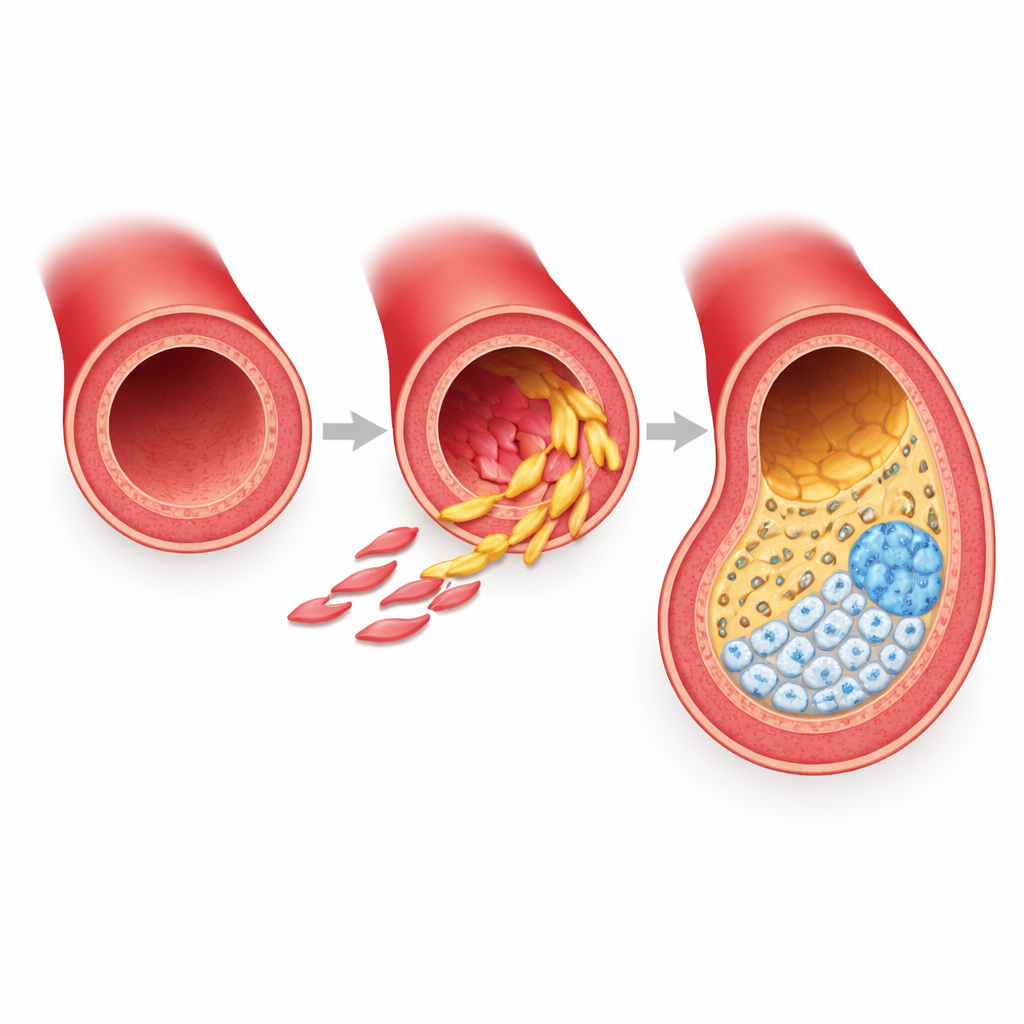
Artery muscle cells that refuse to stay in one role
The work focuses on vascular smooth muscle cells, the contractile cells that form the muscular wall of arteries. Rather than being static structural elements, these cells prove remarkably flexible. Under the stress of a high-fat diet, they abandon their usual job and shift into new identities inside developing plaques. The team used a powerful combination of single-cell RNA sequencing, single-cell chromatin accessibility profiling, and spatial imaging in a standard mouse model of atherosclerosis. This allowed them to watch tens of thousands of individual cells over multiple time points and map where each cell type sits within the artery wall and plaque.
Two fibrous states and a calcified endpoint
By tracking lineage-marked smooth muscle cells only, the researchers identified six related states, with three still strongly resembling contractile muscle and three clearly altered. Two of these altered states are fibrous, called here FMC-1 and FMC-2, and one is calcified and cartilage-like, called CMC. FMC-1 cells appear first, cluster mainly in the middle muscle layer and fibrous cap, and express genes linked to inflammation and immune-like stress responses. FMC-2 cells build up later, concentrate in the fibrous cap and the inner plaque, and are rich in genes for extracellular matrix, collagen organization, wound healing, and lipid handling. Both fibrous states can give rise to the calcified CMC cells at the base of the plaque, which express bone and cartilage programs and correspond to regions of hardening that can destabilize lesions.
Cell fate maps and the gene switches behind them
To turn these snapshots into moving pictures, the team used computational "trajectory" methods that infer how likely each cell state is to change into another over time. They found that one contractile state (SMC-2) is the main starting point that flows into FMC-1 and FMC-2, which in turn feed the calcified CMC state. Importantly, FMC-1 and FMC-2 do not simply disappear; they persist and appear to be sustained fates in mature plaques, rather than brief waystations. By combining RNA expression, chromatin accessibility, and network modeling, the researchers highlighted key transcription factors—master gene switches—that drive these transitions. Factors such as TCF21, ZEB2, SMADs, RUNX family members, and TEAD1 emerge as central players orchestrating when and how muscle cells turn into fibrous or calcified plaque cells.
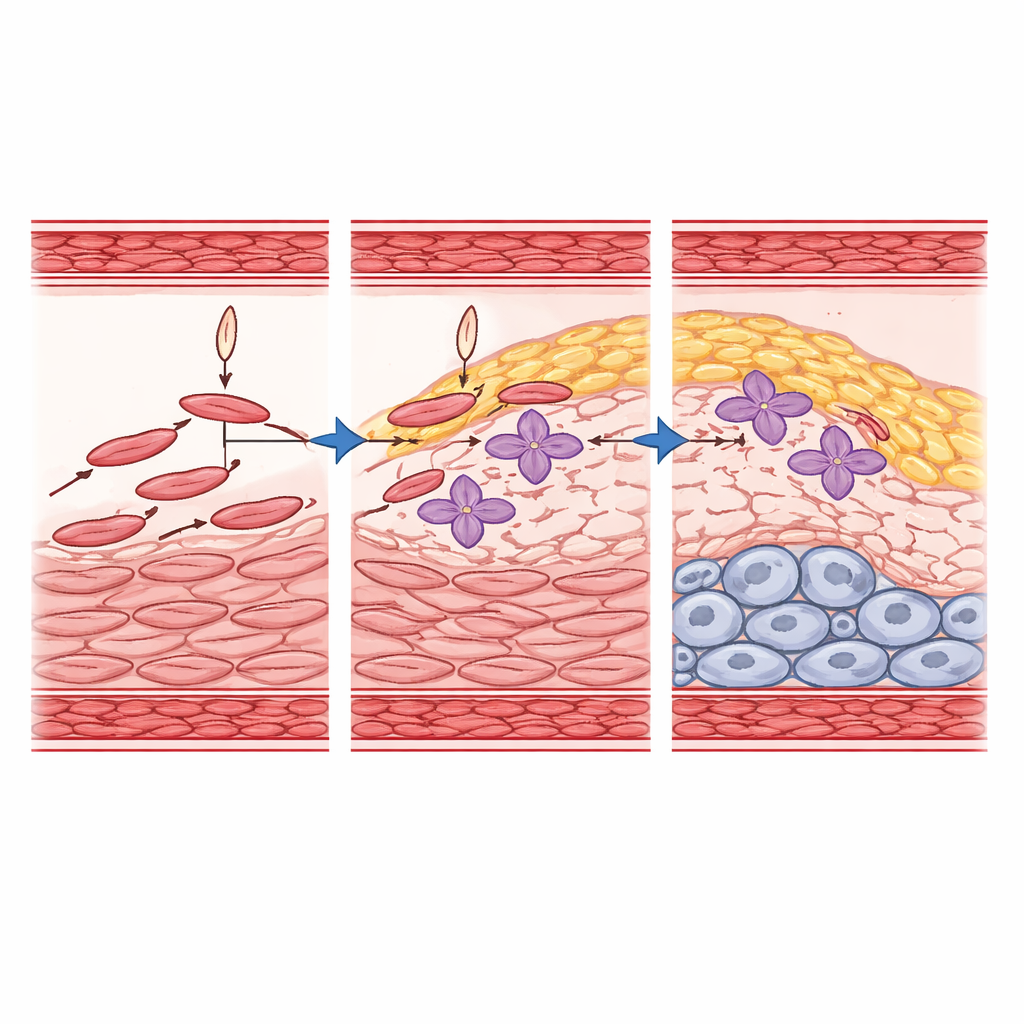
A protective gene that reshapes risky pathways
Among the gene switches, TCF21 stands out because human genetic studies had already linked it to lower risk of coronary artery disease. Using mice in which Tcf21 was deleted specifically in smooth muscle cells, the authors showed that losing this factor reduced the number of fibrous transition cells and strongly blunted progression toward the calcified CMC state. At the same time, other muscle states derived from a particular developmental region of the heart wall expanded, revealing a previously underappreciated compartment under TCF21’s control. Integrating their mouse data with human genome-wide association results, they found that many known risk genes sit in the regulatory networks controlled by TCF21 and its partners, especially TEAD1. These factors physically interact on DNA, fine-tuning the activity of enhancers near genes involved in tissue remodeling, inflammation, and vessel structure.
Linking cell stories to inherited heart risk
To make the jump from mechanism to human disease, the researchers overlaid their single-cell atlas with large-scale human genetic data. They used statistical tools to ask which smooth muscle states are most enriched for genes carrying coronary disease risk variants. One fibrous state, FMC-2, stood out: it harbors a dense concentration of risk-linked genes, some apparently promoting disease and others offering protection. In contrast, the calcified CMC state did not show strong direct enrichment, suggesting that the genetic dice are largely rolled earlier, as cells choose their fibrous fates. Overall, the study paints coronary disease as a problem of misrouted smooth muscle cell trajectories, governed by an interconnected gene network in which TCF21 and co-factors bias cells toward more stable, protective fibrous roles rather than destructive calcified outcomes.
Citation: Li, D.Y., Kundu, S., Cheng, P. et al. Vascular smooth muscle cell state trajectories mediate molecular mechanisms of coronary disease risk. Nat Commun 17, 4059 (2026). https://doi.org/10.1038/s41467-026-70530-z
Keywords: coronary artery disease, smooth muscle cells, atherosclerosis, cell state transitions, TCF21