Clear Sky Science · en
DNA-PK-mediated phosphorylation of STAT6 establishes a non-canonical type 2 immunity axis to prevent macrophage senescence
Why keeping immune cells young matters
Aging bodies often simmer with low-level inflammation, a background “noise” linked to frailty, heart disease, lung problems, and many other conditions. A major culprit is the gradual aging of immune cells called macrophages, which patrol tissues and clear threats and debris. When these sentinels grow old and damaged, they stop repairing themselves properly, spew inflammatory signals, and fail to clean up dying or senescent cells. This study uncovers a built‑in safety switch inside macrophages that helps them repair their DNA, resist aging, and keep tissues healthier for longer.
A hidden line of defense inside immune cells
The researchers focused on how macrophages respond when their DNA is harmed—a common event during aging, pollution exposure, and disease. They discovered that a DNA‑sensing enzyme called DNA‑PK teams up with a well‑known immune regulator, STAT6, in an unexpected way. Instead of only reacting to allergy‑type signals, STAT6 is chemically modified by DNA‑PK at a specific spot (an amino acid called serine 807 in mice). This tiny chemical tag acts as an on–off switch for a protective program: it keeps STAT6 from being broken down, allowing it to stay active longer and redirect the cell toward DNA repair rather than runaway inflammation.
How the switch protects against damage and cell aging
Once stabilized by this modification, STAT6 moves into the nucleus and partners with another protein, PU.1, that shapes the identity of macrophages. Together, they turn on key DNA repair genes, including ones also known from cancer biology such as BRCA1. In mouse macrophages engineered to mimic constant activation of this STAT6 switch, DNA breaks were fixed more efficiently, and classic signs of cellular aging—like accumulation of DNA damage, loss of division capacity, and buildup of waste pigments—were markedly reduced. In contrast, when the serine 807 site was disabled so STAT6 could not be modified, macrophages showed poor DNA repair, entered senescence more readily, and produced strong inflammatory secretions.
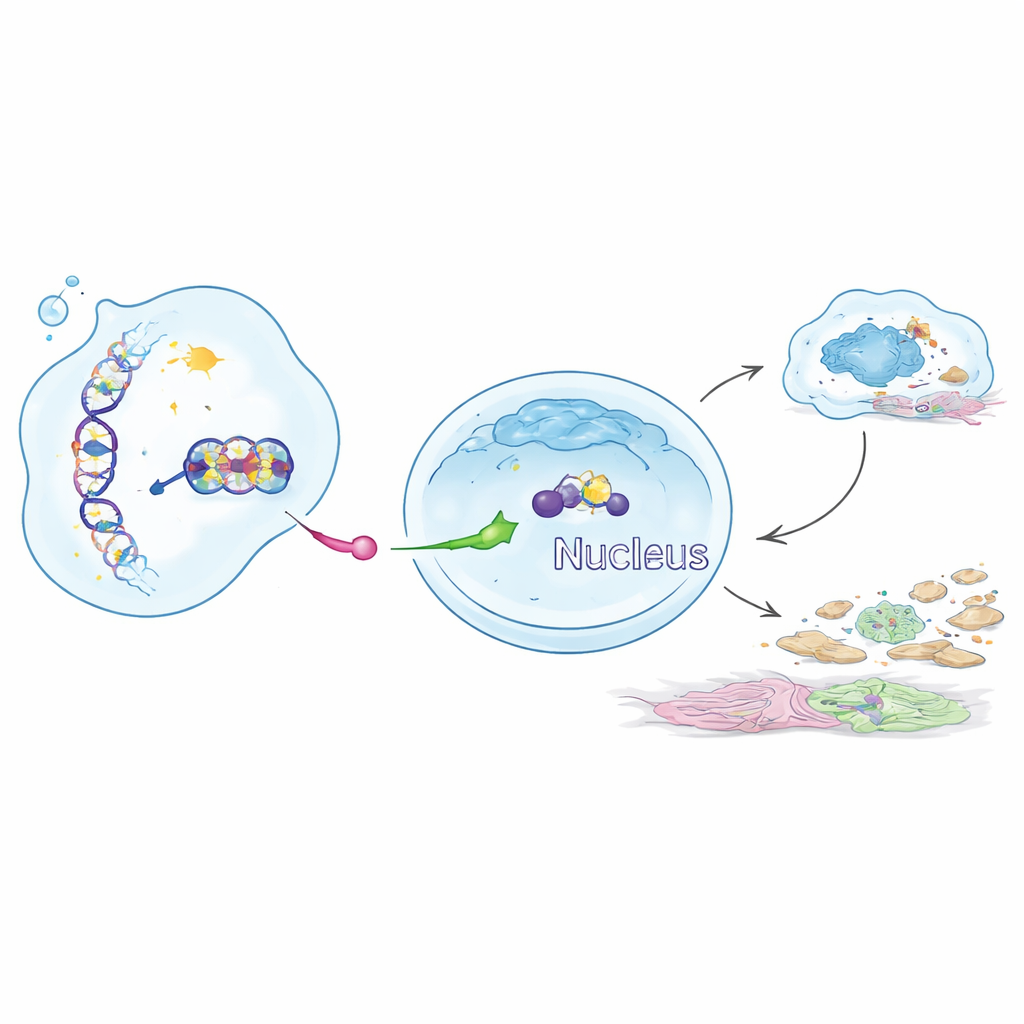
From cell aging to whole‑body decline
The consequences of this microscopic switch extended to the entire organism. Mice that could not modify STAT6 at serine 807 developed more senescent macrophages in multiple organs, had stiffer and more fibrotic tissues, and showed classic aging symptoms: weaker muscles, poorer endurance, reduced bone density, and cognitive decline. Their macrophages were also less able to engulf bacteria or clear senescent cells, undermining immune surveillance. Conversely, mice with the “always‑on” version of the switch were more resilient: their tissues had fewer aging markers, and they performed better on physical and memory tests. Remarkably, transferring macrophages carrying this protective STAT6 form into fast‑aging or naturally old mice improved their physical performance and reduced molecular signs of tissue aging.
Links to lung disease in humans
The team then asked whether a similar mechanism exists in people. Human STAT6 carries a matching site, serine 817, which was shown to be modified by DNA‑PK in human macrophages after DNA damage. As in mice, this modification boosted DNA repair, curbed senescence features, and improved the cells’ ability to engulf particles. Examining lung tissue from patients with chronic obstructive pulmonary disease (COPD)—a common, age‑linked lung condition—they found fewer macrophages with modified STAT6 and more evidence of DNA damage and cell aging. Lower levels of this STAT6 tag strongly correlated with higher markers of senescence, suggesting that failure of this pathway may help drive chronic inflammation and scarring in COPD lungs.
What this means for healthy aging
Taken together, the study reveals a previously unrecognized self‑protection circuit in macrophages: DNA damage activates DNA‑PK, which tags STAT6, stabilizing it and enabling a partnership with PU.1 that turns on DNA repair programs. This keeps macrophages functional, limits their drift into a pro‑inflammatory, senescent state, and helps tissues resist fibrosis and age‑related decline. In both mice and humans, weakening of this circuit is linked to tissue aging and lung disease. Because each step in the pathway involves defined molecules, it offers attractive targets for future therapies—such as drugs or engineered cells designed to boost STAT6’s protective modification—to promote healthier aging and combat chronic inflammatory diseases.
Citation: Zhou, Z., Li, X., Wang, Y. et al. DNA-PK-mediated phosphorylation of STAT6 establishes a non-canonical type 2 immunity axis to prevent macrophage senescence. Nat Commun 17, 3123 (2026). https://doi.org/10.1038/s41467-026-69996-8
Keywords: macrophage senescence, DNA repair, STAT6, inflammaging, COPD