Clear Sky Science · en
Tumour specific HORMAD1 expression perturbs mitotic arrest and drives sensitivity to mitotic kinase inhibitors
Why this matters for cancer treatment
When our cells divide, they rely on intricate safety checks to pass on the right set of chromosomes. Cancer often hijacks or weakens these safeguards, leading to chaotic genomes that both drive disease and shape how tumours respond to drugs. This study uncovers how an unusual protein called HORMAD1, normally active only in reproductive cells, is switched on in many aggressive breast cancers and other tumours. By subtly sabotaging a key cell-division checkpoint, HORMAD1 makes cancer cells more unstable—but also unusually vulnerable to a new class of experimental medicines.
A misplaced fertility protein in cancer cells
HORMAD1 usually appears only in germ cells, where eggs and sperm are made. There, it helps manage DNA reshuffling and quality control during the special type of cell division called meiosis. The authors show that in about 60% of triple-negative breast cancers—and in subsets of several other tumour types—this protein is inappropriately switched back on. Using both engineered non-cancerous cells and cancer cell lines, they found that extra HORMAD1 disrupts the even separation of chromosomes during ordinary cell division. Cells expressing HORMAD1 developed more lagging chromosomes, extra or missing chromosomes (aneuploidy), and small DNA-containing “micronuclei,” all hallmarks of genomic chaos seen in aggressive cancers.
How cell division safety checks normally work
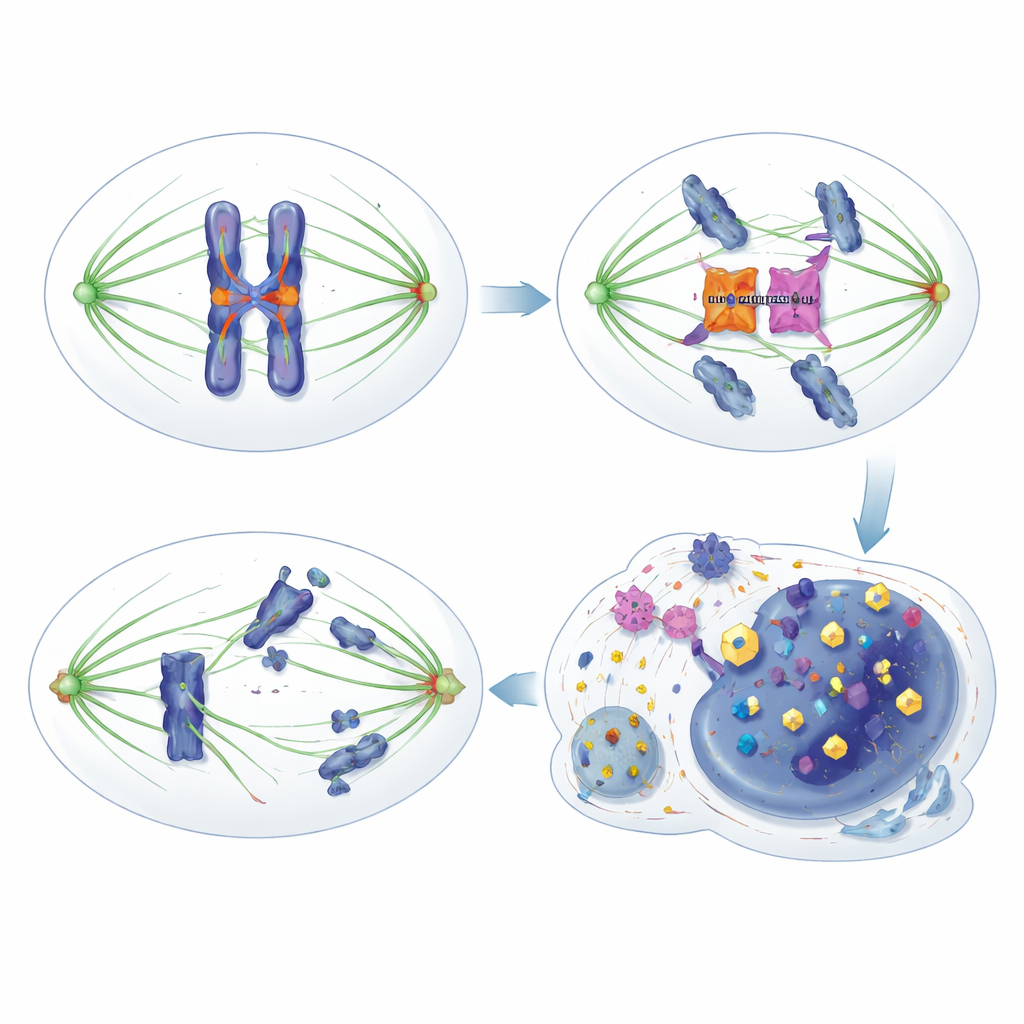
To divide correctly, a cell builds a spindle of microtubule fibers that attach to each chromosome. A surveillance system known as the spindle assembly checkpoint acts like a tension-sensitive brake: if any chromosome is not properly attached, the brake blocks progression, preventing separation until errors are fixed. Several enzymes called mitotic kinases, including MPS1, Aurora B, and BUB1, help sense bad attachments and promote “error correction” so each daughter cell receives the right chromosome set. Disruption of this system can both fuel cancer development and create special weaknesses that certain drugs can target.
HORMAD1 quietly weakens the brake
The researchers discovered that HORMAD1 undermines this safety brake in a subtle but important way. Rather than interfering with classic checkpoint components like the HORMA protein MAD2L1, HORMAD1 binds directly to the kinase Aurora B. Aurora B normally partners with another protein, INCENP, to become fully active and to modify proteins at centromeres and kinetochores—critical sites on chromosomes where spindle fibers attach. When HORMAD1 is present in dividing tumour cells, it competes with INCENP for access to Aurora B, reducing their partnership and dampening Aurora B’s activity. As a result, Aurora B’s usual phosphorylation signals on several targets are weakened, error correction is less effective, and the checkpoint becomes “leaky”: cells exit mitosis too soon, even when attachments are faulty, leading to mis-segregated chromosomes and genomic instability.
From weakness to therapeutic opportunity
Because HORMAD1 only partially disables Aurora B and related safeguards, cancer cells remain just viable enough to grow, but depend heavily on the remaining function of mitotic kinases to survive repeated faulty divisions. The team tested this by exposing HORMAD1-positive and HORMAD1-negative cells to experimental inhibitors of MPS1, Aurora B, and BUB1. Across multiple models, HORMAD1 expression made cells far more sensitive to these drugs, dramatically reducing their ability to proliferate or form colonies. Depleting BUB1 genetically was especially lethal only in the presence of HORMAD1, revealing a strong, selective dependency. In mouse models using patient-derived triple-negative breast tumours, those with high HORMAD1 levels shrank or grew more slowly when treated with a nanoparticle formulation of an Aurora B inhibitor, whereas HORMAD1-negative tumours largely resisted the same treatment.
What this means for patients
To a lay observer, HORMAD1 acts like a double-edged sword in cancer: it pushes tumour cells toward greater chromosomal disorder, which can drive disease, but in doing so it also makes them precariously reliant on a few remaining cell-division safeguards. The study shows that this misplaced fertility protein weakens a key checkpoint by misdirecting Aurora B, leaving HORMAD1-positive tumours particularly vulnerable to drugs that target Aurora B, MPS1, or BUB1. Because HORMAD1 is largely absent from normal tissues yet present in a clear subset of cancers, it could serve as a biomarker to identify patients most likely to benefit from these emerging mitotic kinase inhibitors, potentially opening new targeted treatment avenues for difficult-to-treat cancers such as triple-negative breast cancer.
Citation: Walker, C., Kollarovic, G., Weekes, D. et al. Tumour specific HORMAD1 expression perturbs mitotic arrest and drives sensitivity to mitotic kinase inhibitors. Nat Commun 17, 2157 (2026). https://doi.org/10.1038/s41467-026-69561-3
Keywords: HORMAD1, triple-negative breast cancer, chromosomal instability, Aurora B kinase, mitotic checkpoint inhibitors