Clear Sky Science · en
Complement 5a receptor 2 attenuates diabetic kidney disease by promoting mitochondria-associated endoplasmic reticulum membrane formation mediated by PSS-MFN2 interaction
Why this kidney research matters
Diabetic kidney disease is one of the most common reasons people with diabetes end up needing dialysis or a transplant, and current treatments mainly slow, rather than stop, the damage. This study uncovers an unexpected natural defense system inside kidney cells that helps them cope with the toxic mix of high sugar and fat in diabetes. By revealing how a little‑known immune receptor, C5aR2, protects kidney cells’ energy factories and fat handling, the work points to a new kind of drug that could shield kidneys without shutting down the immune system.
A surprising guardian in the diabetic kidney
The researchers focused on diabetic kidney disease, in which tiny structures in the kidney gradually scar and fail under long‑term exposure to high blood sugar and disturbed metabolism. They examined kidney biopsies from patients and found that a receptor called C5aR2 was strongly increased in the tissue between the kidney filters, especially in proximal tubular cells that reabsorb nutrients. Higher C5aR2 levels went hand in hand with worse disease and a greater chance of progressing to kidney failure, suggesting that the receptor becomes more active as damage accumulates. Curiously, earlier work had framed C5aR2 mainly as a “decoy” immune receptor; this paper shows it also has an important role in cellular metabolism.
When the defender is missing, damage accelerates
To test whether C5aR2 is friend or foe, the team used diabetic mice that lacked the C5ar2 gene. Compared with diabetic mice that still had the receptor, these knockout animals developed heavier protein loss in the urine, more scarring and inflammation in the kidney tissue, and more structural damage seen under the microscope. Their tubular cells were clogged with fat droplets, showed strong signs of stress in the endoplasmic reticulum (the cell’s folding and packaging station), and had swollen, poorly working mitochondria. Similar problems appeared in cultured kidney cells when C5aR2 was silenced, including reduced oxygen consumption, a direct readout of mitochondrial performance. Together, these findings indicate that C5aR2 normally helps tubular cells withstand the metabolic stress of diabetes.
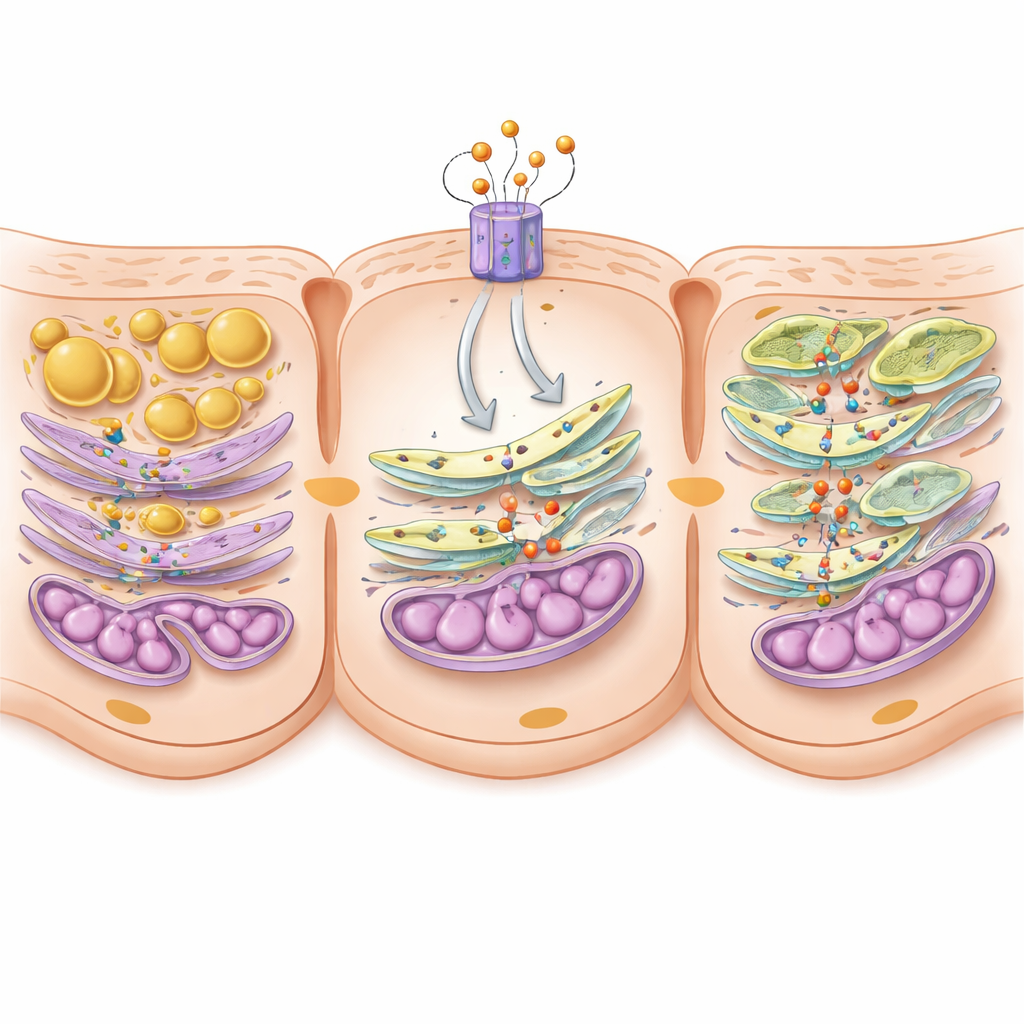
How tiny contact sites and a key fat ingredient keep cells healthy
Digging deeper, the scientists profiled lipids in the kidneys and found that mice without C5aR2 had a marked loss of phosphatidylserine, a crucial building block of cell membranes, while neutral storage fats were increased. Phosphatidylserine is mainly made at specialized junctions where the endoplasmic reticulum and mitochondria touch, called mitochondria‑associated membranes. At these junctions, enzymes named PSS1 and PSS2 synthesize phosphatidylserine, and a tethering protein called MFN2 helps pass it to mitochondria. In diabetic mice, these contact zones were already shortened; removing C5aR2 made them shrink even more, and the amount of PSS1, PSS2, and MFN2 in the junctions dropped. In cells, C5aR2 activity turned out to boost a transcription factor, c‑FOS, which in turn increased production of the PSS enzymes. The team further showed that PSS proteins physically bind MFN2 to form a functional bridge that supports both contact formation and lipid transfer.
Restoring the missing link to rescue stressed cells
To prove that this molecular bridge really matters, the authors artificially raised PSS2 levels only in kidney tubular cells of diabetic mice. Despite ongoing diabetes, these animals had less protein in the urine, less scarring, fewer fat droplets, improved mitochondrial shape and function, and longer contact zones between mitochondria and endoplasmic reticulum. Phosphatidylserine levels in these junctions also bounced back. In cell experiments, boosting PSS2 restored energy production and reduced fat buildup even when C5aR2 was suppressed, confirming that PSS2 sits at a critical point in this protective pathway.
A new drug strategy that tunes, rather than blocks, complement
Because completely blocking complement signaling can impair host defense, the researchers explored a more selective tactic: activating C5aR2 with a designer small peptide called P59. In diabetic db/db mice, P59 given under the skin for 10 weeks reduced body weight and blood triglycerides, lowered urinary protein loss, and markedly improved tubulointerstitial damage. Kidneys from treated mice showed fewer lipid droplets, less endoplasmic reticulum stress, healthier mitochondria, and stronger mitochondria‑associated membranes enriched with PSS1, PSS2, MFN2, and phosphatidylserine. Single‑cell RNA sequencing revealed that P59 specifically revived Pss2 expression in injured proximal tubule cells. In cultured kidney cells, P59’s benefits disappeared when C5aR2 or the PSS enzymes were knocked down, showing that its protective effects travel through this newly mapped C5aR2–c‑FOS–PSS–MFN2 axis.
What this means for people living with diabetes
In everyday terms, this study suggests that kidneys under diabetic stress try to switch on C5aR2 to keep their energy systems and fat handling in order. When this receptor is missing or overwhelmed, tiny contact sites between cellular compartments fall apart, a key membrane ingredient runs low, and fat and stress signals pile up, driving scarring. By gently stimulating C5aR2 with a targeted drug, it may be possible to rebuild these contact bridges, restore healthier lipid balance, and protect kidney function without broadly suppressing the immune system. While much work remains before such treatments reach the clinic, the findings open a promising new avenue to slow or prevent kidney failure in people with diabetes.
Citation: Zhao, Yy., Wang, Yh., Li, Zh. et al. Complement 5a receptor 2 attenuates diabetic kidney disease by promoting mitochondria-associated endoplasmic reticulum membrane formation mediated by PSS-MFN2 interaction. Cell Discov 12, 24 (2026). https://doi.org/10.1038/s41421-026-00873-w
Keywords: diabetic kidney disease, mitochondria-associated membranes, lipid metabolism, complement receptor C5aR2, proximal tubular cells