Clear Sky Science · en
Inhibiting HSP27 activates the XBP1s/CerS1 interplay, which triggers DRP1-driven mitophagy, thereby protecting against cell death and promoting the KSHV lytic cycle in primary effusion lymphoma cells
When Cell Stress Becomes a Double-Edged Sword
Our cells survive daily insults by switching on emergency programs that repair damage and keep them alive. Cancer cells, however, can hijack these same programs to grow and to shelter lurking viruses. This article explores how blocking a single stress‑protective protein in a rare lymphoma not only nudges tumor cells toward death but also gives a hidden virus inside them a window to wake up and multiply. Understanding this delicate balance could help design therapies that kill cancer while denying the virus a chance to spread.
A Hidden Virus in Aggressive Lymphoma
Primary effusion lymphoma is a highly aggressive cancer of B cells, a type of white blood cell. Most of these tumor cells carry a dormant passenger: Kaposi’s sarcoma‑associated herpesvirus (KSHV). In its quiet, or latent, state the virus produces only a few proteins and hides within the host genome. Certain stresses can push it into an active, lytic phase in which it copies itself and produces new viral particles, usually killing the host cell. The tumor cells themselves depend on several stress‑response systems, including so‑called heat shock proteins and the unfolded protein response, which help them cope with misfolded proteins, disturbed fat metabolism, and damage to their energy‑producing mitochondria. 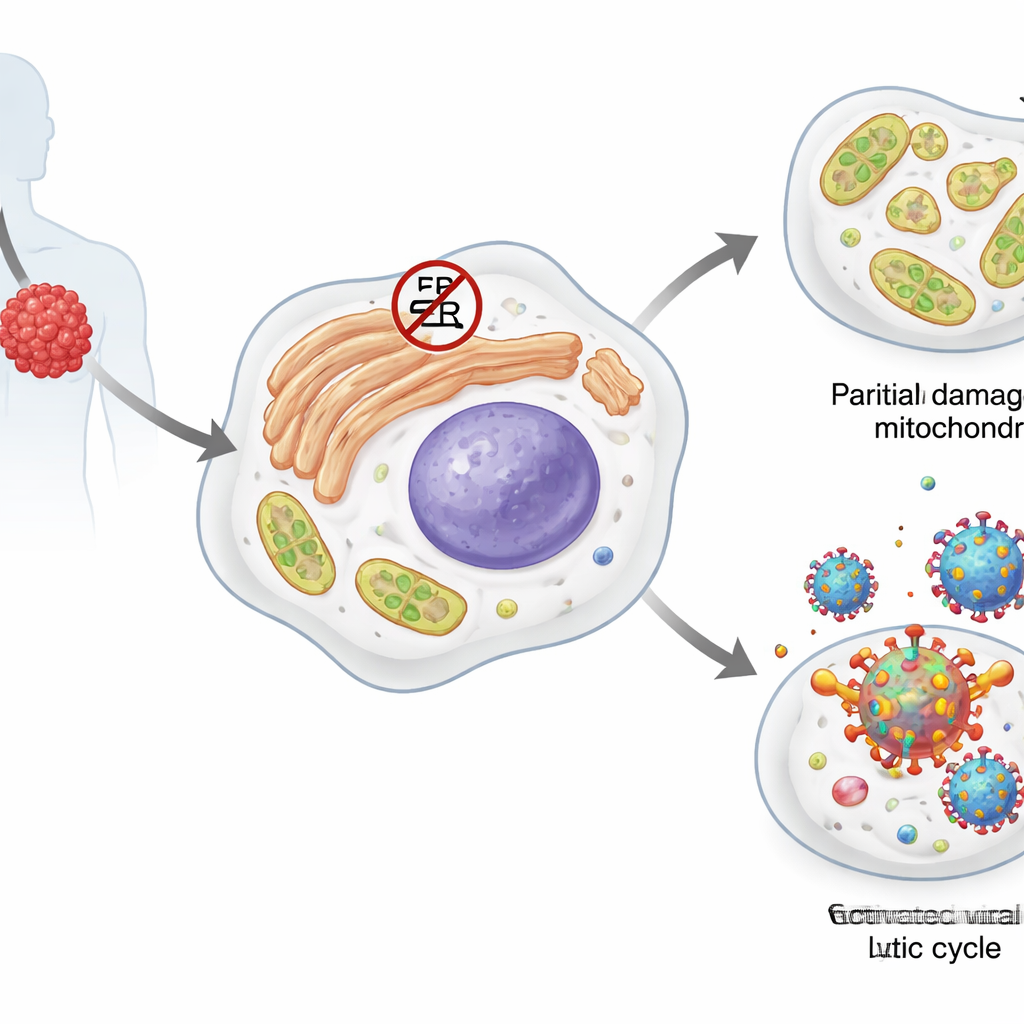
Blocking a Cellular Bodyguard
The researchers focused on HSP27, a small heat shock protein known to shield cells from stress. Using a chemical inhibitor called J2 or by silencing the gene with small RNAs, they reduced HSP27 activity in lymphoma cells growing in the lab. This made the cells less likely to survive and triggered a strong stress signal in an internal membrane network called the endoplasmic reticulum. Markers of this response, including protective and pro‑death factors, rose, and a key switch called XBP1s was turned on. At the same time, the cells showed more signs of programmed cell death, confirming that taking away HSP27 pushes them toward a tipping point between survival and demise.
A Stress Loop That Talks to Cell Fats
Stress in the endoplasmic reticulum is closely intertwined with how cells handle fats. The team found that blocking HSP27 increased levels of CerS1, an enzyme that makes a specific fat molecule called C18‑ceramide. When they chemically blocked XBP1s, the rise in CerS1 disappeared, showing that XBP1s helps turn on the CerS1 gene under these conditions. Strikingly, inhibiting CerS1 in turn lowered XBP1s, revealing a positive feedback loop: each factor supports the other. This molecular handshake not only reshapes fat metabolism but also strengthens the cell’s ability to adapt to endoplasmic reticulum stress, even while death signals are building.
Mitochondria Recycled Instead of Destroyed
Stress in one part of the cell often spills over into the mitochondria, the tiny power plants that generate energy. After HSP27 was blocked, the lymphoma cells produced more reactive oxygen species, a sign of mitochondrial trouble, and increased levels of DRP1, a protein that cuts mitochondria into smaller pieces. The authors showed that the XBP1s–CerS1 loop was responsible for raising DRP1. This, in turn, triggered mitophagy, a quality‑control process in which damaged mitochondria are wrapped in membranes and delivered to cellular “recycling centers” called lysosomes. Using fluorescent dyes and protein markers, they confirmed that mitochondria were being selectively removed. When they chemically or genetically blocked DRP1, this mitophagy was reduced and the cells died more readily, meaning that mitochondrial recycling was actually helping the stressed tumor cells hang on. 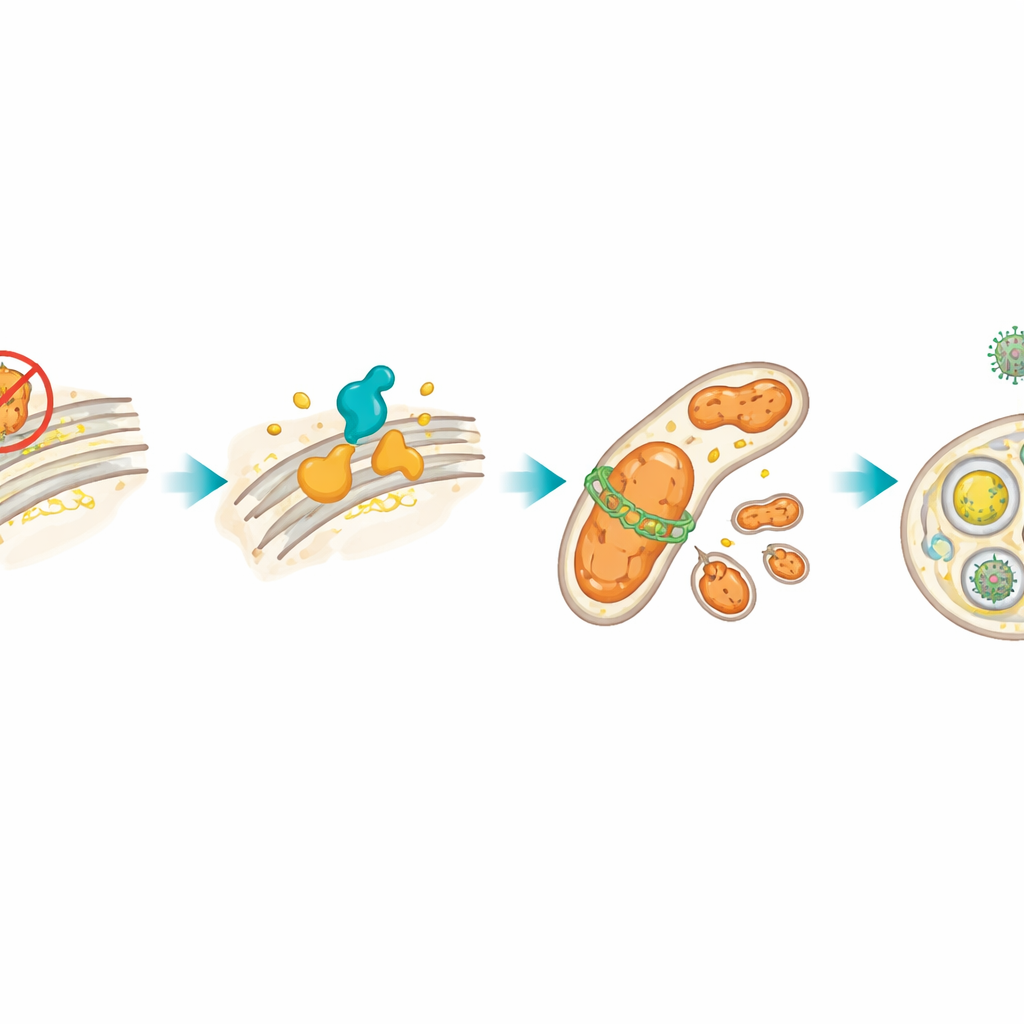
Giving the Virus Time to Escape
The same mitophagy that protected the tumor cells also benefitted KSHV. Activation of XBP1s, accumulation of C18‑ceramide, and increased mitochondrial fission have all been linked to reawakening this virus. Here, when HSP27 was inhibited, more cells expressed early and late viral proteins, clear signs of lytic replication. Blocking DRP1, and therefore mitophagy, reduced this viral reactivation. The authors suggest that by slightly prolonging cell survival under stress, mitophagy gives KSHV time to complete its replication cycle and leave the dying cell, potentially infecting new targets and contributing to cancer development.
What This Means for Future Treatments
To a non‑specialist, the key message is that HSP27 acts as a central traffic controller for how lymphoma cells cope with stress, how they recycle damaged mitochondria, and how a cancer‑linked virus decides when to wake up. Turning off HSP27 unleashes a chain of events that both undermines cell survival and, paradoxically, temporarily protects cells through mitophagy while allowing KSHV to replicate. Therapeutically, combining HSP27 inhibition with drugs that block DRP1‑driven mitophagy could push tumor cells to die faster and limit the virus’s chance to spread, offering a two‑pronged strategy against this deadly lymphoma.
Citation: Gonnella, R., Corrado, V., Scaffidi, G.F. et al. Inhibiting HSP27 activates the XBP1s/CerS1 interplay, which triggers DRP1-driven mitophagy, thereby protecting against cell death and promoting the KSHV lytic cycle in primary effusion lymphoma cells. Cell Death Discov. 12, 118 (2026). https://doi.org/10.1038/s41420-026-02979-2
Keywords: primary effusion lymphoma, Kaposi sarcoma virus, cell stress response, mitophagy, heat shock protein HSP27