Clear Sky Science · en
A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder
Why this mouse study matters for inflammation
Many severe illnesses, from deadly infections to autoimmune flare-ups, are driven not just by germs or genes, but by the body’s own runaway inflammation. A protein called RIPK3 has long been seen as a key executioner of a violent form of cell death that fuels such inflammation, making it an attractive drug target. But RIPK3 also has other, less understood roles inside cells. This study describes a new kind of lab mouse that cleanly separates RIPK3’s killing activity from its signaling “support” role, revealing how each contributes to inflammation and pointing to new therapeutic strategies.
Two ways a death protein can act
Cells can die in orderly or messy ways. In tidy, “silent” cell death, the body quietly recycles cell parts without much alarm. In a messier form called necroptosis, cells burst open and spill their contents, triggering strong immune responses. RIPK3 is central to necroptosis: when turned on, it activates another protein that punches holes in the cell membrane. However, earlier work suggested RIPK3 can also help trigger more classical, caspase-driven cell suicide and can boost inflammatory signaling even without killing cells. Teasing apart these roles has been difficult because existing inactive forms of RIPK3 either kill embryos or drastically lower the protein’s levels, making it hard to study its normal scaffolding behavior.
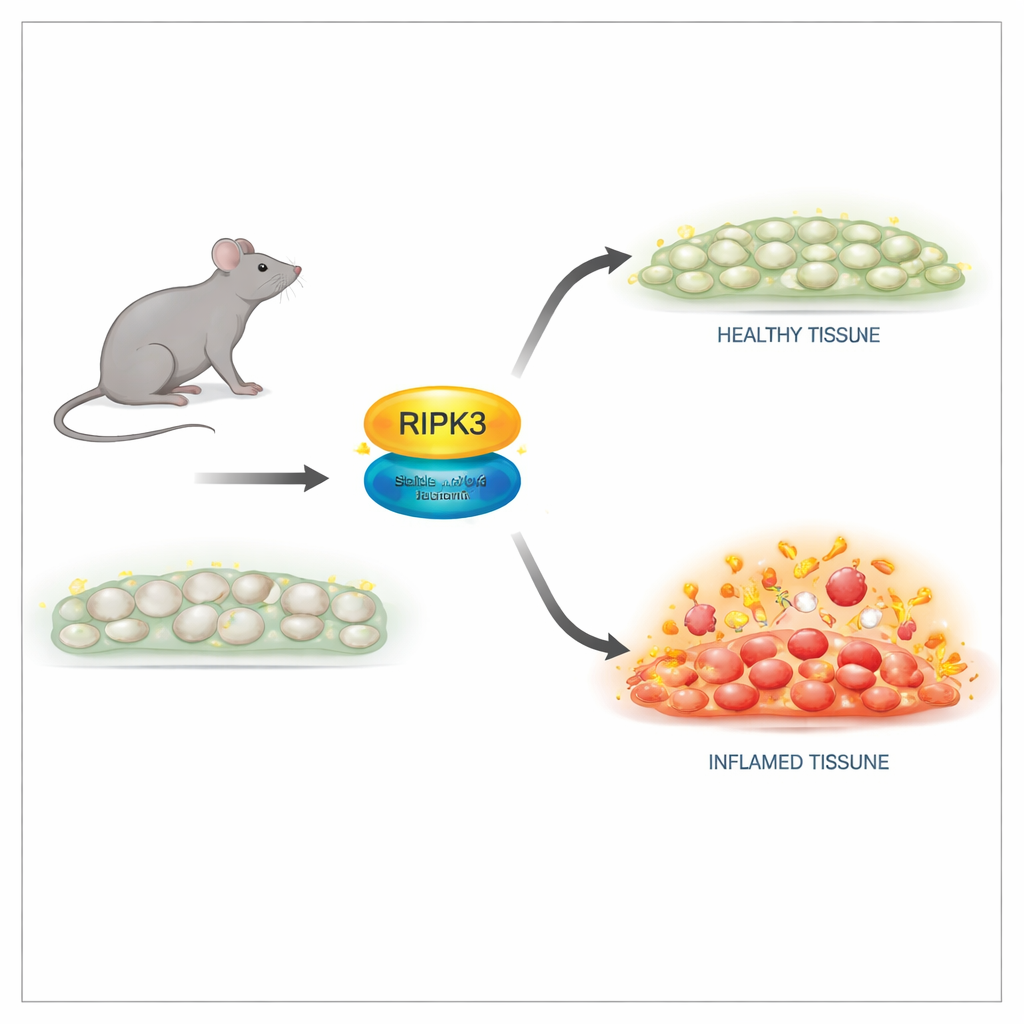
A safer way to switch off the killing function
The researchers engineered mice carrying a subtle change in the RIPK3 protein at a single position, termed D143N, that shuts down its enzyme activity while preserving its structure. In cells from these mice, RIPK3 protein levels and tissue architecture looked normal, and the animals were born and grew just like their healthy littermates. Importantly, cells with the D143N version were completely resistant to multiple necroptosis triggers, including signals from tumor necrosis factor (TNF), toll-like receptors, and viral infection. The mutant RIPK3 could no longer activate its downstream partner or form the destructive complex needed for membrane rupture, yet it did not provoke spontaneous apoptosis, avoiding the lethal side effects seen in older RIPK3 mutants.
Separating development from disease
One of RIPK3’s best-known roles is in embryos that lack another key protein, caspase-8: without caspase-8, RIPK3-driven necroptosis kills the embryo. In this study, introducing the D143N version of RIPK3 completely rescued these otherwise nonviable mice. They developed normally and were fertile, proving that RIPK3’s killing activity is dispensable for normal development when its structure is preserved. Yet when adult mice were challenged with high-dose TNF to induce a shock-like inflammatory syndrome, the picture changed. Animals completely lacking RIPK3 were strongly protected from death, tissue damage, and inflammatory molecules in the blood. Mice with the D143N version, despite lacking necroptosis, were only partly protected. This indicated that RIPK3’s non-killing, scaffold role still helped drive inflammation.
Scaffold signaling that fans the flames
To understand this non-lethal contribution, the team examined gene activity in the gut of TNF-treated mice. In RIPK3-deficient animals, many inflammatory genes were strongly dampened. In D143N mice, however, the suppression was weaker, and genes linked to interferon and innate immune responses remained more active. At the protein level, TNF robustly turned on the JAK–STAT1 and ERK signaling pathways in normal and D143N mice, but this activation was almost completely absent when RIPK3 was deleted entirely. This showed that even without its killing function, RIPK3’s physical presence in signaling complexes helps transmit TNF signals into a pro-inflammatory program via JAK–STAT1.
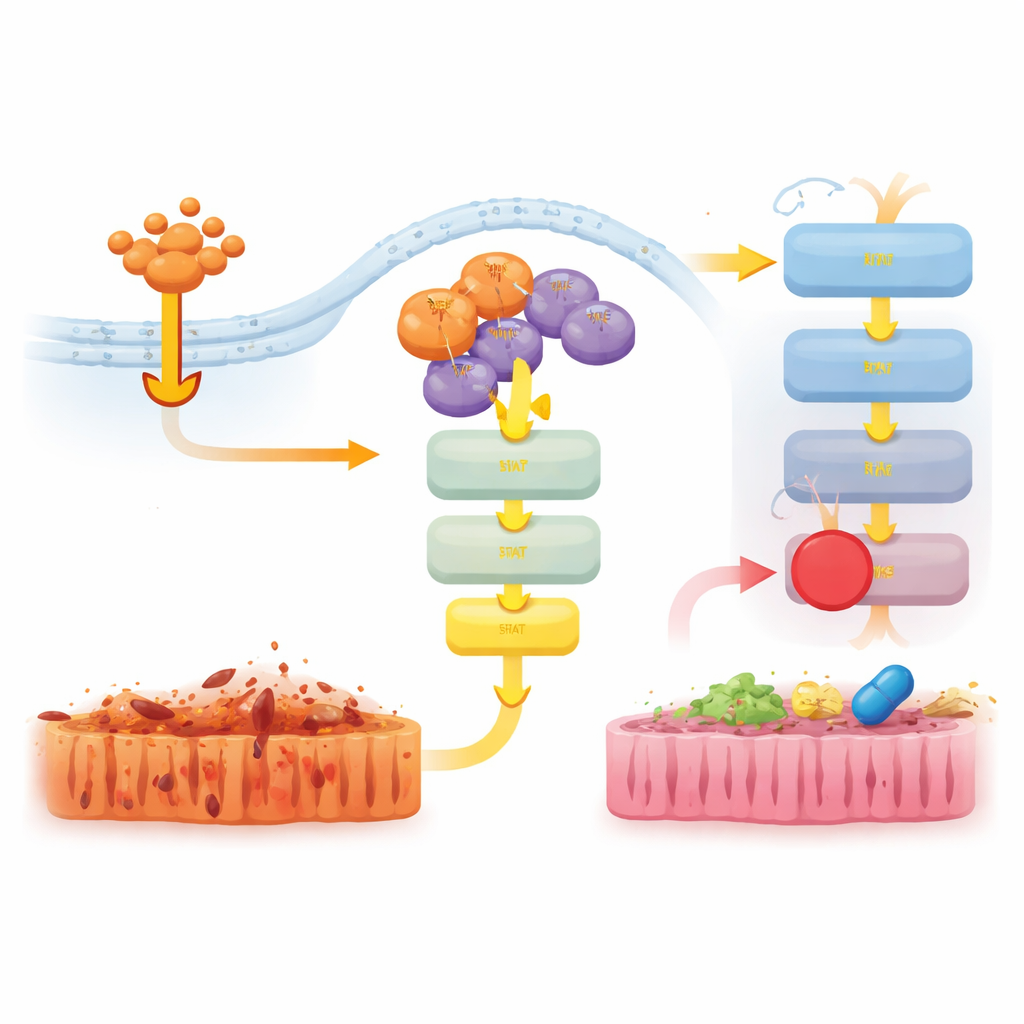
Turning down harmful signals with targeted drugs
The researchers then tested whether blocking these downstream pathways could ease disease in D143N mice undergoing TNF-induced shock. Treating the animals with a JAK1/2 inhibitor, but not an ERK inhibitor, reduced body temperature loss, lowered levels of the inflammatory molecule IL-6, and lessened gut tissue damage and cell death. A separate inhibitor targeting another protein, RIPK1, also strongly protected the mice and dampened JAK–STAT1 and ERK activation. Together, these results suggest that RIPK3’s scaffold function teams up with RIPK1 to activate JAK–STAT1 and drive inflammation, and that interrupting this signaling can reduce tissue injury even when necroptosis is already blocked.
What this means for future treatments
For years, RIPK3 has been viewed mainly as a switch for a toxic form of cell death, and drug development has focused on shutting off its enzyme activity. This study shows that doing so may not be enough: RIPK3 can still act as a physical platform that amplifies inflammatory signals through JAK–STAT1, contributing to shock and tissue damage. The new D143N mouse model reveals these dual roles with unusual clarity, establishing a powerful tool to study when and how each function matters in different diseases. For patients, the work suggests that combining RIPK3- or RIPK1-targeting drugs with blockers of JAK–STAT1 could more effectively calm harmful inflammation in conditions driven by TNF and related cytokines.
Citation: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
Keywords: RIPK3, necroptosis, inflammation, TNF shock, JAK-STAT1