Clear Sky Science · en
GDPD5-CD55-EGFR competitive binding axis regulates radioresistance and lipid accumulation in rectal cancer
Why treatment sometimes falls short
For people with rectal cancer, radiation given before surgery is meant to shrink tumors and lower the chance that the disease returns. Yet many tumors stubbornly resist this treatment. This study explores an unexpected culprit in that resistance: how cancer cells handle fats inside their own walls, and how a small group of surface proteins together tip the balance between cells that die after radiation and cells that survive.
When cancer cells stockpile fat
Researchers first compared tissue samples from rectal cancer patients who responded well to pre-surgery chemoradiotherapy with those whose tumors barely shrank. Using gene data from dozens of patients and computer analysis of hundreds of fat-related genes, they found a clear pattern: tumors that resisted radiation tended to load up on fat droplets. In cell and mouse models, rectal cancer cells made resistant in the lab also showed more fat storage, higher levels of a fat-building enzyme and lower levels of a fat-burning enzyme. When the team forced ordinary cancer cells to build more fat, the cells became harder to kill with radiation, while blocking fat production made resistant cells easier to damage.
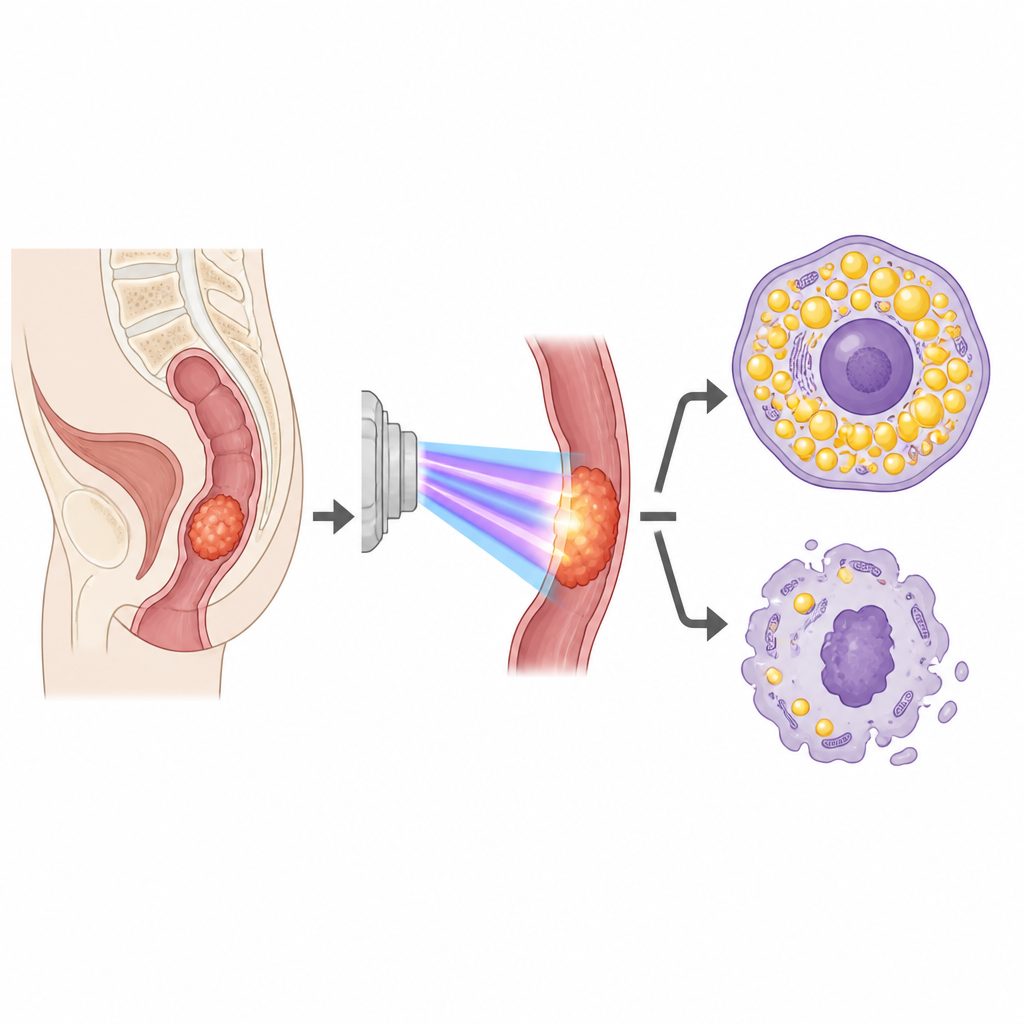
A key switch on the cell surface
To move beyond this pattern and find what drives it, the scientists used several machine-learning methods to sift through the fat-related genes tied to treatment response. One gene, called GDPD5, stood out as the strongest predictor of whether a tumor would resist chemoradiotherapy. Resistant cell lines had much higher GDPD5 levels than their original counterparts. When the team silenced GDPD5 in these resistant cells, fat stores inside the cells shrank, the fat-building enzyme dropped, the fat-breaking enzyme rose, and the cells became more sensitive to radiation. Re-adding GDPD5 reversed these effects. In mice, tumors formed from GDPD5-silenced cells grew more slowly after radiation, carried fewer fat deposits, and had fewer actively dividing cells.
How a three-part protein chain shields the cell
Digging into the mechanism, the researchers focused on p53, a well-known guardian protein that helps cells either repair radiation damage or self-destruct. Genetic analysis showed that turning down GDPD5 activated the p53 pathway, and earlier work has linked p53 to both fat use and radiation response. When the team knocked down p53 at the same time as GDPD5, the benefits disappeared: fat re-accumulated and radiation once again killed fewer cells. They then asked how GDPD5 might be tampering with p53. Attention turned to EGFR, a growth receptor that, when it slips into the cell nucleus, can weaken p53’s function. Resistant cells showed more EGFR inside their nuclei, but this nuclear presence dropped when GDPD5 was silenced.
The tug-of-war for a growth receptor
Biochemical tests revealed that GDPD5 does not grab EGFR directly. Instead, both proteins bind to a third partner on the cell surface called CD55. Computer docking and pull-down experiments suggested that GDPD5 and EGFR compete for the same spots on CD55. When CD55 binds EGFR, it helps anchor the receptor at the cell membrane, limiting its journey into the nucleus. When GDPD5 levels are high, it displaces EGFR from CD55, freeing EGFR to travel inward. In cell and animal models, removing GDPD5 reduced nuclear EGFR and boosted p53 activity, but removing CD55 at the same time restored EGFR’s nuclear entry, fat buildup, and radiation resistance. In patient samples and three-dimensional organoids grown from patient tumors, high GDPD5 paired with low CD55 went hand in hand with more EGFR in nuclei and poorer responses to radiation.
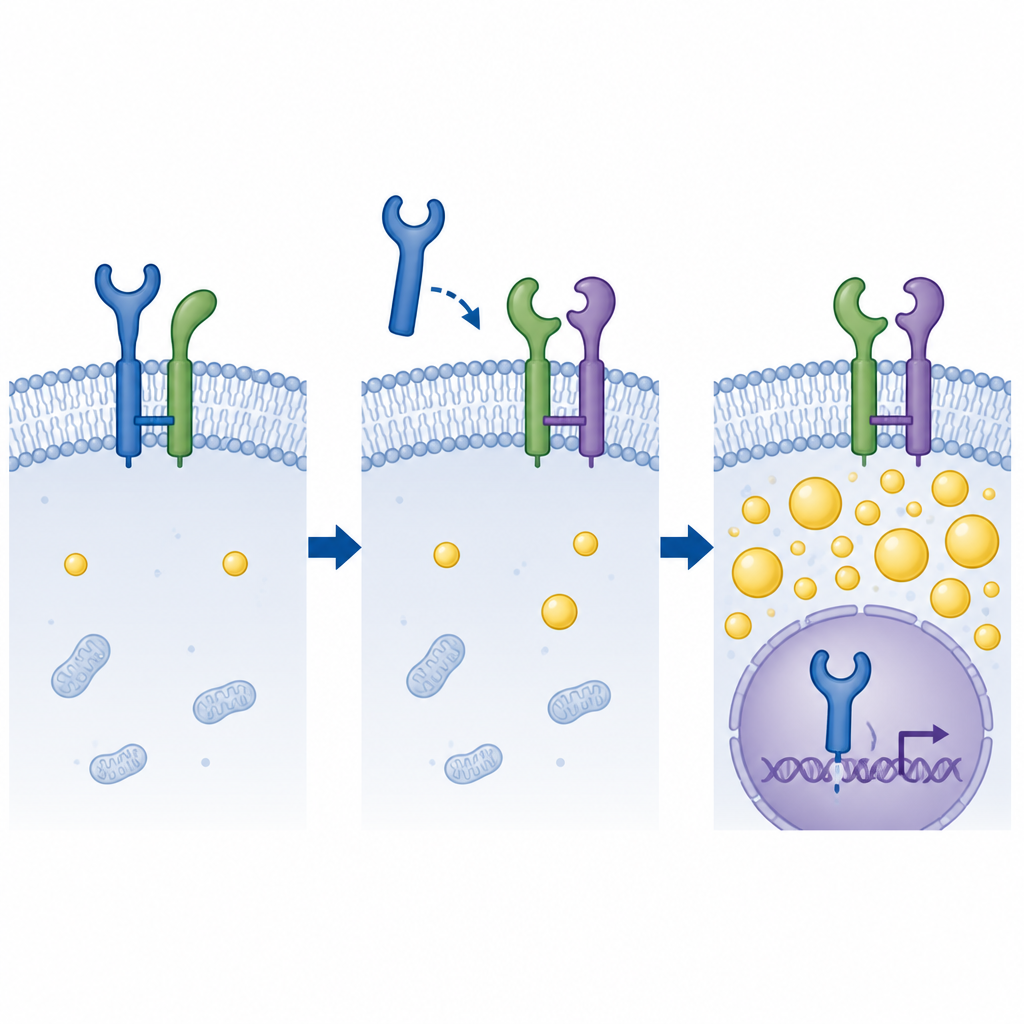
What this means for patients
Put simply, the study shows that a protein called GDPD5 helps rectal cancer cells survive radiation by breaking a protective link between CD55 and EGFR on the cell surface. This change lets EGFR move into the nucleus, where it quiets p53, encourages fat hoarding, and makes cells harder to kill. Tumors with high GDPD5 and low CD55 are more likely to resist treatment, while dialing down GDPD5 or restoring CD55’s hold on EGFR makes cells more vulnerable to radiation. Although these findings are still at the laboratory and early organoid stage, they point to a new strategy: drugs that block the GDPD5–CD55–EGFR interaction might one day help standard radiotherapy work better for people with rectal cancer.
Citation: Zhu, R., Li, M., Shen, Y. et al. GDPD5-CD55-EGFR competitive binding axis regulates radioresistance and lipid accumulation in rectal cancer. Cell Death Dis 17, 492 (2026). https://doi.org/10.1038/s41419-026-08711-3
Keywords: rectal cancer, radioresistance, lipid metabolism, EGFR signaling, p53 pathway