Clear Sky Science · en
AKR1C3–PKM2–oxidative phosphorylation axis drives prostate cancer radioresistance via UBE2T upregulation
Why some prostate tumors shrug off radiation
Radiation therapy is a mainstay treatment for prostate cancer, yet in many men the tumor eventually comes back. This study asks a simple question with big consequences: what makes some prostate cancer cells withstand radiation that kills others? By tracing how these cells fuel themselves and repair their DNA, the researchers uncover a molecular "power line" that helps tumors survive treatment and suggest a new target that could make radiotherapy work better.
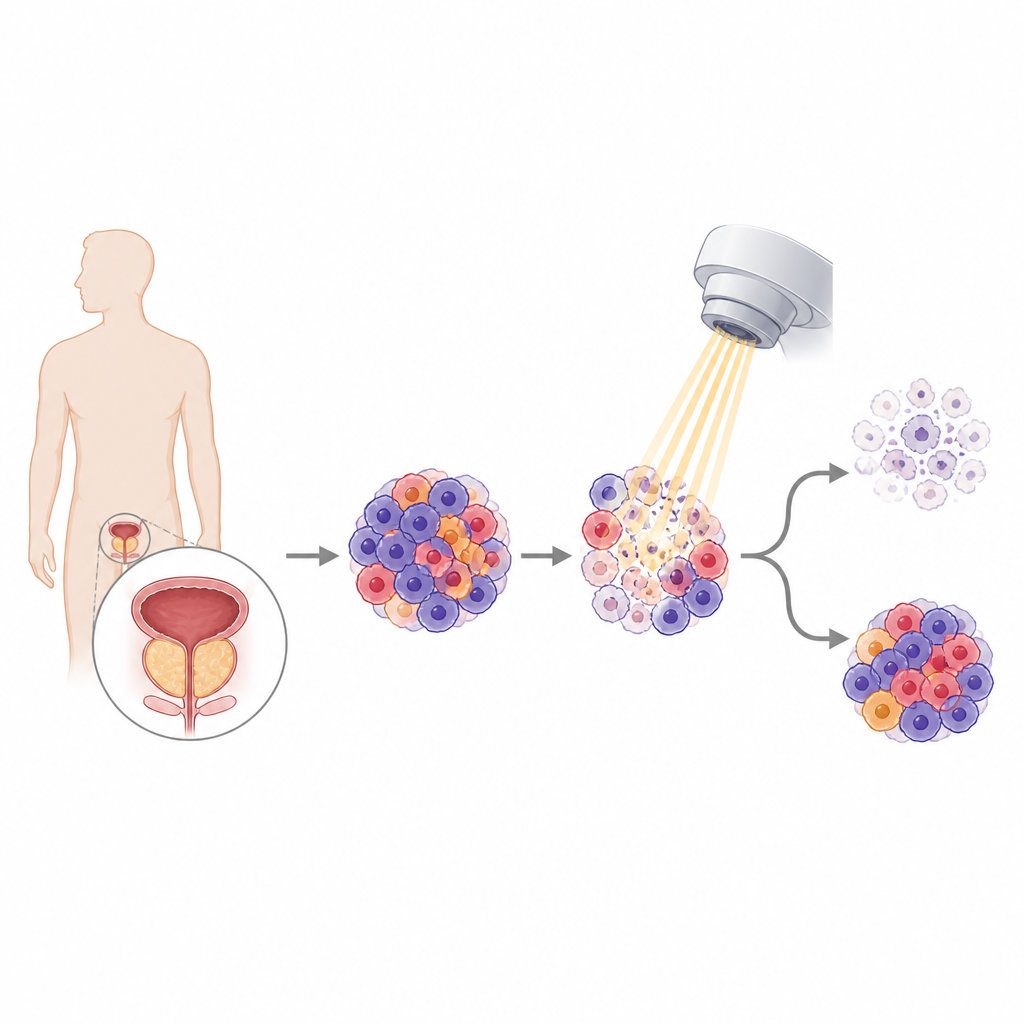
A search through patient data for hidden culprits
The team began by mining large public databases of prostate cancer samples from patients who had received radiotherapy. They compared gene activity in men whose tumors responded well with those whose tumors resisted treatment, and also in early versus advanced disease. Thirteen genes rose to the top as being linked to both poor response to radiation and progression to aggressive, treatment-resistant cancer. One gene, called AKR1C3, stood out because higher levels were tied to shorter periods without the disease returning and more advanced tumor stage, marking it as a strong suspect in radioresistance.
Putting the suspect gene to the test in cancer cells
To see whether AKR1C3 actually helps prostate cancer cells survive radiation, the researchers manipulated its levels in several cell lines grown in the lab. When they boosted AKR1C3, cells formed more colonies after radiation and showed less DNA damage. When they reduced AKR1C3, the opposite happened: radiation left more damage and fewer cells survived. These effects appeared not only in cells that rely on male hormone signals but also in cells that do not, suggesting AKR1C3 protects tumors through an additional route beyond well-known hormone pathways.
A metabolic switch that powers DNA repair
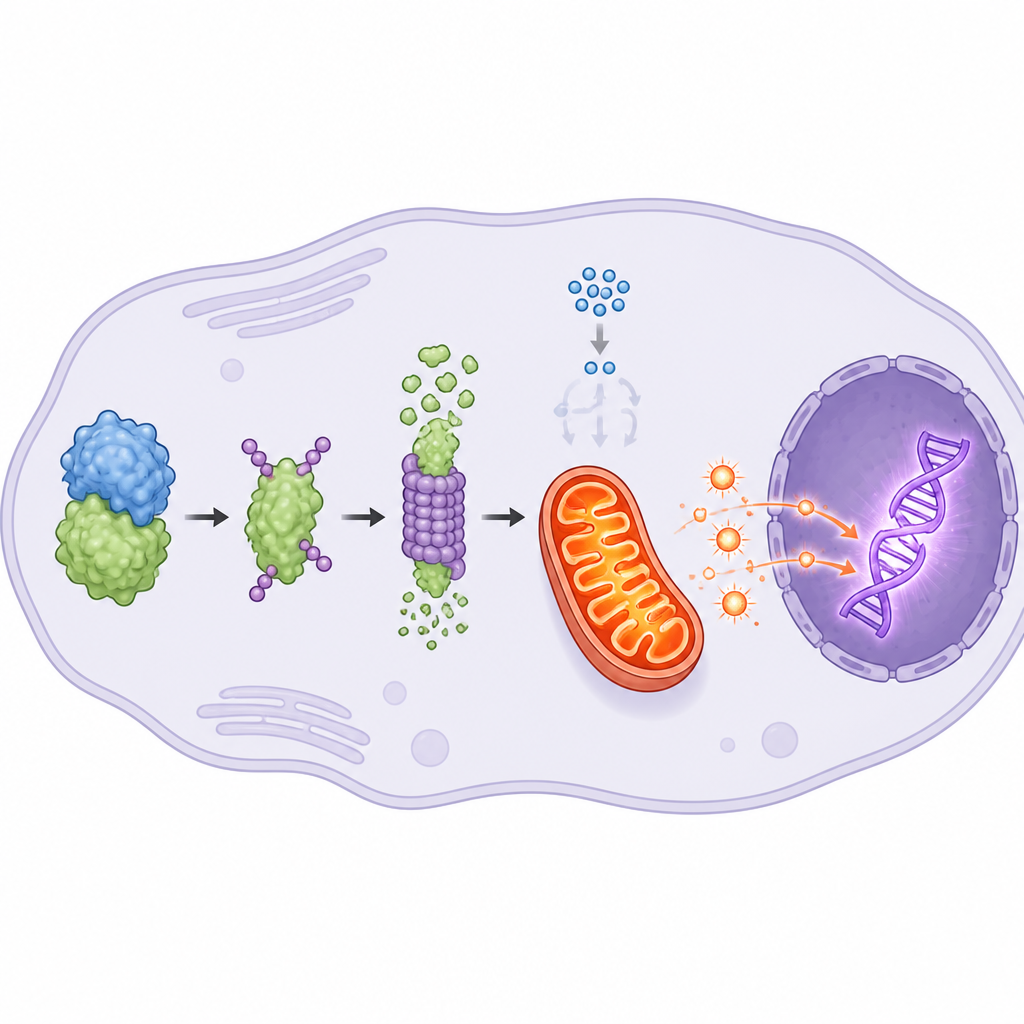
Diving deeper, the group found that AKR1C3 changes how cancer cells make and use energy. Rather than relying mainly on quick sugar burning, cells with high AKR1C3 shift toward a more efficient process in the cell’s power plants, the mitochondria. AKR1C3 binds to and speeds the breakdown of another enzyme, PKM2, which normally drives glycolysis, the classic sugar-hungry route favored by many tumors. With PKM2 reduced, cells lean more on oxidative phosphorylation, a mitochondrial process that yields more energy and modestly raises levels of reactive oxygen molecules. This extra energy supplies DNA repair machinery, while the chemical by-products spur a sensor protein, NRF2, to move into the nucleus and switch on repair genes, especially one called UBE2T that is strongly tied to resistance to radiation.
From cell dishes to mice: testing a potential weak point
The scientists then confirmed this chain of events in mice carrying human prostate tumors. When they lowered AKR1C3 in tumors and gave radiation, the tumors shrank more and showed more signs of DNA damage than with radiation alone. Conversely, tumors forced to make extra AKR1C3 were harder to control. The team also tried a drug, ASP9521, that fits into AKR1C3’s active pocket. This blocker both weakened the interaction between AKR1C3 and PKM2 and reduced levels of the repair helper UBE2T. In mice, combining the drug with radiation led to smaller tumors and more tumor cell death than either treatment by itself, without obvious harm to liver or kidney tissues.
What this means for future prostate cancer treatment
In plain terms, the study reveals a wiring diagram inside prostate cancer cells where AKR1C3 flips a metabolic switch, rerouting energy production through mitochondria to better fuel DNA repair after radiation. By helping break down PKM2, AKR1C3 boosts mitochondrial output, raises chemical signals that activate NRF2 and ultimately increases UBE2T, a key repair helper. This chain of events allows tumor cells to patch up radiation damage and keep growing. Targeting AKR1C3 with selective drugs, especially alongside radiotherapy, could make resistant tumors more vulnerable and may help delay or prevent progression to more lethal forms of prostate cancer.
Citation: Zhang, J., Li, J., Yan, Y. et al. AKR1C3–PKM2–oxidative phosphorylation axis drives prostate cancer radioresistance via UBE2T upregulation. Cell Death Dis 17, 433 (2026). https://doi.org/10.1038/s41419-026-08666-5
Keywords: prostate cancer, radiotherapy resistance, tumor metabolism, DNA repair, AKR1C3