Clear Sky Science · en
STING activation induces polarized cytokine secretion of IFN-β and IL-17A promoting photoreceptor death and choroidal disruption in age-related macular degeneration
Why this matters for aging vision
Age-related macular degeneration (AMD) is a leading cause of irreversible vision loss in older adults, robbing people of the ability to read, drive, and recognize faces. Yet doctors still lack treatments that reliably halt the dry, atrophic form of the disease. This study uncovers a central alarm system inside support cells of the retina that, when chronically triggered, appears to coordinate much of the damage seen in AMD. Understanding this master switch offers a path toward therapies that might protect both light-sensing cells and the blood vessels that nourish them.
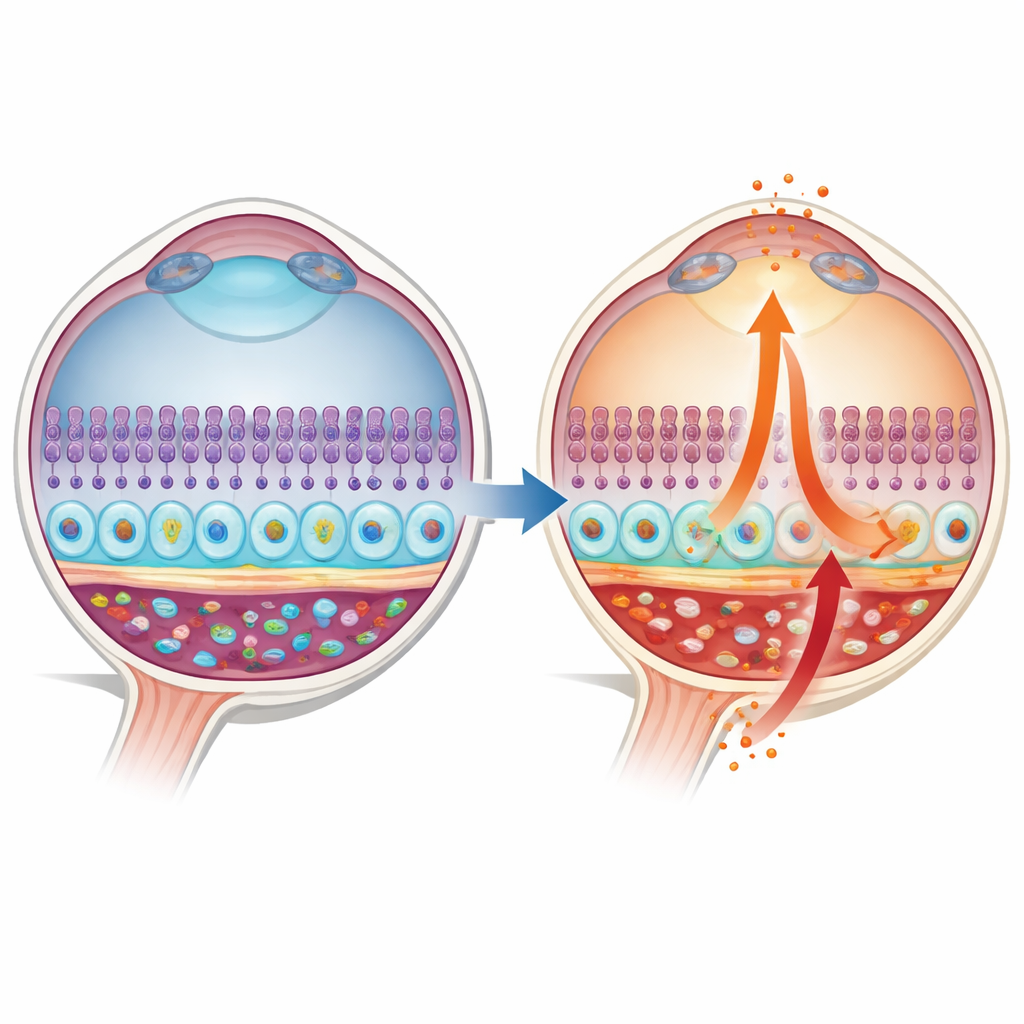
A hidden alarm in the eye’s support layer
The macula sits at the center of the retina and depends on a thin support sheet called the retinal pigment epithelium (RPE). On one side, RPE cells face the photoreceptors that capture light; on the other, they face a rich bed of blood vessels in the choroid. The authors focused on a molecular alarm pathway known as STING, which normally detects stray DNA inside cells and briefly switches on antiviral defenses. In donated human eyes, they found that STING and one of its key messengers, interferon‑beta (IFN‑β), were low and confined to a narrow zone in healthy RPE. In contrast, in eyes with early and late AMD, STING became much more abundant and spread throughout the RPE cell body, with IFN‑β levels rising in parallel, suggesting a shift from quiet surveillance to chronic alarm.
Two opposing flows of inflammation
To understand how this alarm damages vision, the team grew RPE cells in the lab from induced pluripotent stem cells derived from siblings, where one had AMD and the other did not. They cultured these cells on porous membranes that preserve their natural top‑and‑bottom orientation. RPE from AMD patients released far more IFN‑β from their top (apical) side, the surface that faces photoreceptors. When this apical fluid was applied to human retinal organoids—mini‑retina spheres grown from stem cells—it triggered marked photoreceptor death, an effect that could be mimicked by adding IFN‑β directly. At the same time, the same AMD‑derived RPE secreted much higher levels of another inflammatory messenger, IL‑17A, from their bottom (basal) side toward the choroidal vessels, while apical IL‑17A stayed low. This “polarized” pattern offers a concrete explanation for how one cell layer can simultaneously injure photoreceptors and destabilize the underlying blood supply.
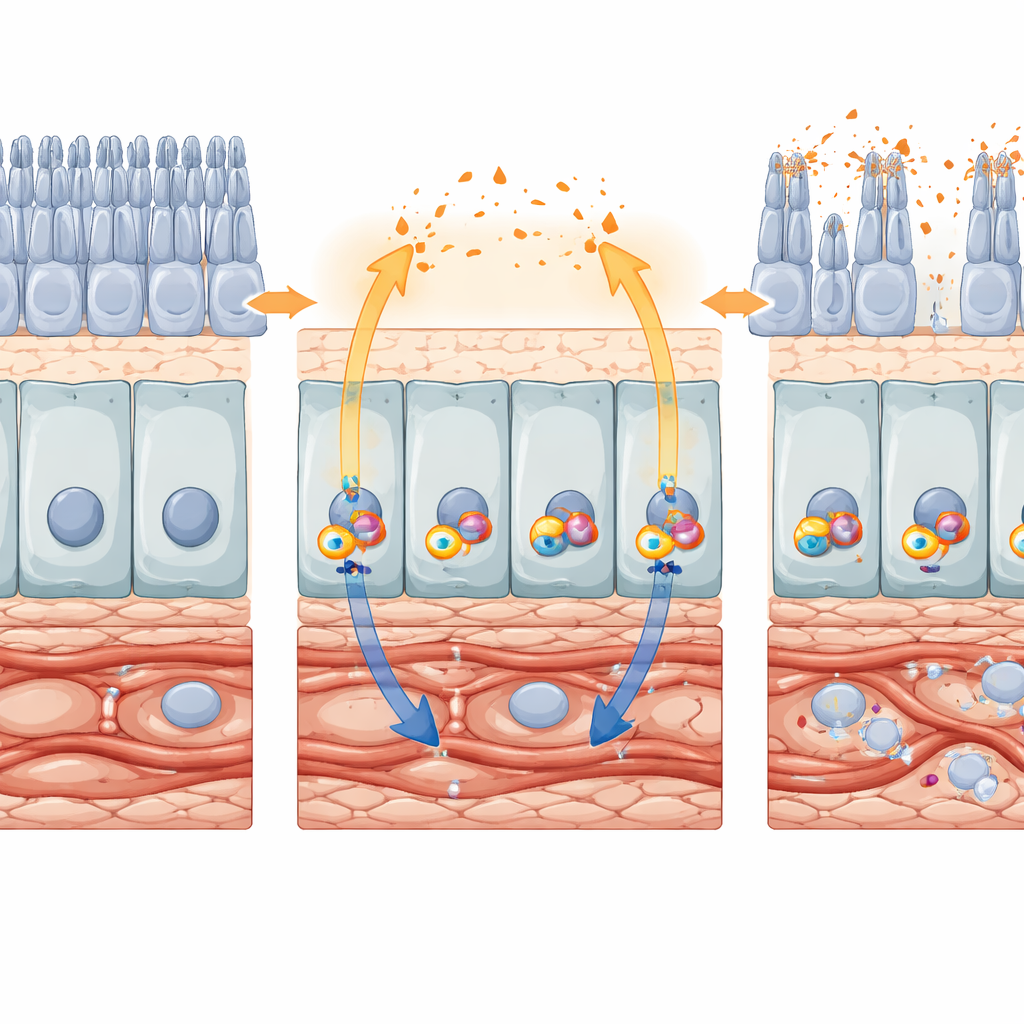
From protective signal to destructive loop
Animal models confirmed that this polarized signaling is not just a cell‑culture curiosity. In mice lacking a protein called Cryba1 in the RPE—an AMD‑like model with waste‑clearing defects—the authors observed increased STING activity and strong upregulation of IL‑17A, accompanied by thickened choroids, stressed RPE, and shortened photoreceptor segments. Separate experiments forced long‑term IFN‑β production in mouse eyes using a viral vector. After ten weeks, gene‑expression profiling showed broad activation of interferon responses and disruption of visual and metabolic genes in RPE and retina. By twenty weeks, imaging and microscopy revealed widespread RPE atrophy, photoreceptor loss, vascular regression, and signs of cellular aging and senescence. Together, these results support a feed‑forward loop: STING activates IFN‑β, IFN‑β boosts IL‑17A (directly and via other interferons), and these signals reinforce chronic inflammation and tissue degeneration.
Zooming in on the retina’s energy crisis
At the single‑cell level, long‑lasting IFN‑β signaling reshaped how retinal cells handle energy. Rod photoreceptors, which normally rely heavily on sugar breakdown (glycolysis), dialed down glycolytic genes and ramped up mitochondrial energy‑production genes. This shift may leave them more vulnerable to stress in an already energy‑hungry tissue. Support cells called Müller glia also showed altered pathways tied to metabolism and nerve protection. Meanwhile, RPE cells exposed to IFN‑β in culture and in vivo lost key identity markers and tight junctions, and upregulated classic senescence markers such as p16 and p21. Senescent RPE not only function poorly but also secrete their own cocktail of inflammatory factors, adding fuel to the degenerative fire started by STING.
Switching off the alarm to protect sight
Encouragingly, the study also shows that dialing down this pathway can rescue retinal structure and function. A reversible small‑molecule STING inhibitor, SN‑011, strongly reduced IFN‑β, IL‑17A, and other cytokines in human RPE and improved their mitochondrial performance. In mice, partially reducing STING in Cryba1‑deficient animals lowered IL‑17A, preserved RPE architecture, and improved electrical responses to light compared with full Cryba1 knockouts. These benefits persisted with age, suggesting that early, targeted interference with STING can slow or prevent the cascade that leads to geographic atrophy. For patients, the work positions STING as a unifying target that links waste‑clearance problems, mitochondrial stress, and runaway inflammation in AMD, raising the prospect of treatments that protect both photoreceptors and their life‑support system rather than tackling each problem separately.
Citation: Huang, C., Babu, V.S., Bammidi, S. et al. STING activation induces polarized cytokine secretion of IFN-β and IL-17A promoting photoreceptor death and choroidal disruption in age-related macular degeneration. Cell Death Dis 17, 283 (2026). https://doi.org/10.1038/s41419-026-08491-w
Keywords: age-related macular degeneration, retinal inflammation, STING pathway, interferon beta, retinal pigment epithelium