Clear Sky Science · en
Short-chain acyl-CoA dehydrogenase initiates mtDNA demethylation and leakage to fuel antitumor immunity in colorectal cancer
Why our own cells sometimes hide cancer from the immune system
Colorectal cancer is one of the world’s deadliest tumors, in part because the body’s immune defenses often fail to recognize and attack it. This study uncovers an unexpected link between how cancer cells burn fats, how their tiny power plants (mitochondria) handle their DNA, and whether the immune system is alerted to the tumor’s presence. By tracing this chain of events, the researchers also highlight an old natural compound, hypericin, as a potential way to reawaken immune attack in colorectal cancer.
A missing mitochondrial “guardian” in colon tumors
The team began by searching large human and mouse datasets to find metabolic genes that change consistently in colorectal cancer. One enzyme stood out: short-chain acyl-CoA dehydrogenase, or ACADS, which normally helps mitochondria break down short fatty acids. In both patient samples and several mouse models, ACADS levels were markedly lower in tumor tissue than in nearby healthy colon. When the scientists reduced ACADS in mouse colon cancer cells, tumors grew faster and more aggressively; boosting ACADS slowed tumor growth. Mice engineered to lack ACADS specifically in their intestinal lining developed more and larger tumors in a chemical model of colitis-associated cancer, supporting the idea that ACADS acts as a tumor suppressor in the gut.
How tumors turn down immune alarm signals
These growth effects could not be explained just by how quickly cancer cells multiplied in a dish, which changed little. Instead, ACADS loss only drove tumor growth in animals with intact immune systems, pointing to changes in the tumor microenvironment. Single-cell analyses of human colorectal cancers showed that tumors with low ACADS were surrounded by more tumor cells and suppressive immune cells—such as myeloid-derived suppressor cells, certain macrophages, and regulatory T cells—and fewer helpful T cells and natural killer cells. This pattern points to an “immunosuppressive neighborhood” that shelters the cancer from attack. 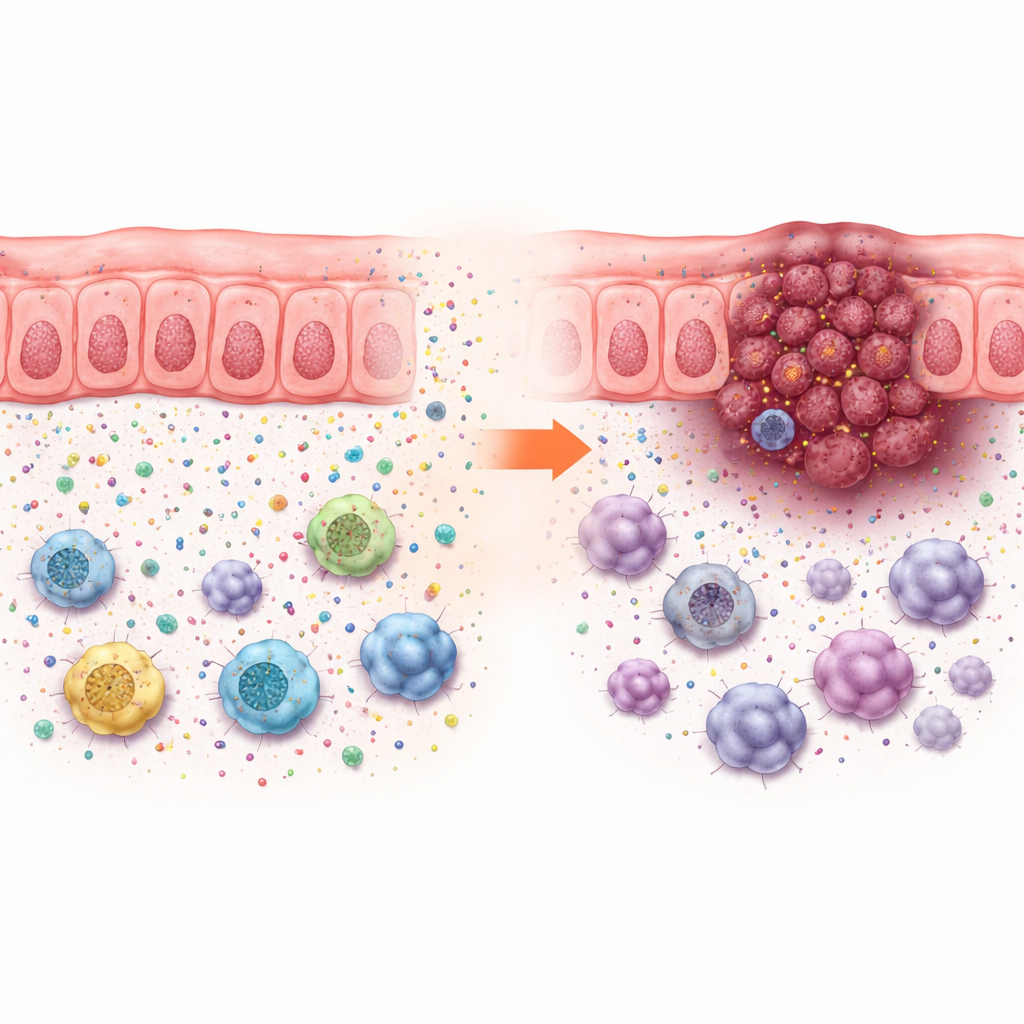
Mitochondrial DNA leakage as the hidden trigger
What connects a fat-burning enzyme to an immune DNA sensor? The answer lies in mitochondrial DNA (mtDNA). Under stress, fragments of mtDNA can leak from mitochondria into the surrounding fluid of the cell, where cGAS detects them as a danger signal. The researchers showed that ACADS-deficient cancer cells had less mtDNA in this compartment, even though total mtDNA was unchanged. Blocking mtDNA leakage in ACADS-high cells shut down cGAS–STING, confirming that these escaped DNA fragments are the critical alarm. Surprisingly, classic mitochondrial stress factors such as reactive oxygen species, calcium surges, and major changes in mitochondrial shape could not fully explain the difference. Instead, the study points to the “gates” in the mitochondrial membrane and, more importantly, to the chemical marks on the mtDNA itself. 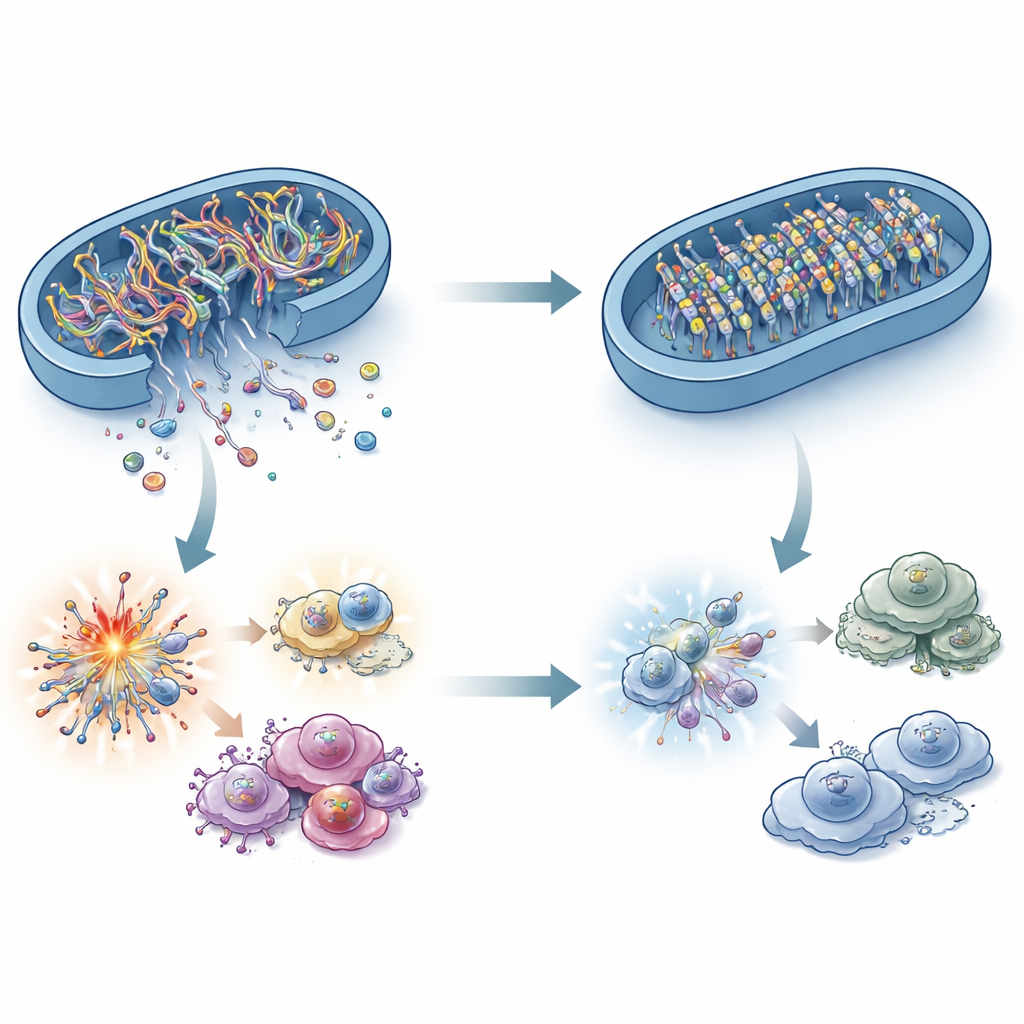
A DNA-methylating partner that locks the alarm inside
Through protein interaction screens, ACADS was found to associate with a form of the DNA methylation enzyme DNMT1 that localizes to mitochondria. When ACADS was lost, this mitochondrial DNMT1 accumulated, placing extra methyl groups on mtDNA. These marks make mtDNA more stable and less prone to breakage and leakage. Overloading cells with mitochondrial DNMT1 reduced mtDNA escape, dampened cGAS–STING signaling, and accelerated tumor growth, while blocking DNMT1 with the drug decitabine restored mtDNA leakage and slowed ACADS-deficient tumors. Patient samples mirrored these findings: low ACADS aligned with high mitochondrial DNMT1, weaker STING signaling, fewer effector T cells, more suppressive immune cells, and poorer predicted responses to checkpoint immunotherapy.
Reawakening immune defenses with an old compound
To see if this pathway could be exploited therapeutically, the researchers used computer-based screening to search for molecules that bind ACADS. They identified hypericin, a natural pigment previously tested as a light-activated treatment for certain skin lymphomas. In colorectal cancer cells, hypericin increased ACADS levels, reduced mitochondrial DNMT1, boosted mtDNA leakage, and reactivated cGAS–STING signaling—changes that depended on the presence of ACADS. In mouse tumor models and in short-term cultures of human colorectal tumors, hypericin treatment shrank tumors or shifted immune cells toward a more active, T-cell–rich state. Although further work is needed before clinical use, these results suggest that pharmacologically “turning back on” ACADS may help convert a cold, immunosuppressed tumor into one that responds better to immunotherapy.
What this means for patients and future treatments
In everyday terms, this work shows that some colorectal cancers grow in part because they silence a mitochondrial enzyme that normally helps leak tiny snippets of DNA into the cell’s interior, where they act as flares to call in the immune system. By letting a DNA-methylating partner lock that mitochondrial DNA in place, ACADS-deficient tumors keep those flares hidden and avoid immune detection. Restoring ACADS activity, for example with hypericin-like drugs, could reopen this mitochondrial alarm system, strengthen antitumor immunity, and improve responses to existing immunotherapies. ACADS, mitochondrial DNMT1, and STING pathway activity may therefore serve as useful biomarkers and targets in the quest for more effective colorectal cancer treatments.
Citation: Yang, F., Wang, M., Hu, S. et al. Short-chain acyl-CoA dehydrogenase initiates mtDNA demethylation and leakage to fuel antitumor immunity in colorectal cancer. Sig Transduct Target Ther 11, 113 (2026). https://doi.org/10.1038/s41392-026-02675-8
Keywords: colorectal cancer, tumor immunity, mitochondrial DNA, lipid metabolism, cGAS-STING pathway