Clear Sky Science · en
Targeting fused in sarcoma (FUS): a novel antisense strategy for treating idiopathic pulmonary fibrosis
Why Scarring Lungs Matter
Idiopathic pulmonary fibrosis (IPF) is a relentless lung disease in which delicate air sacs slowly turn into stiff scar tissue, making every breath harder. Today’s drugs can slow this scarring but cannot stop or reverse it. This study explores a new target called FUS, a protein that helps cells handle their genetic messages, and tests whether switching it off with a designer strand of DNA-like material could calm the scarring process and help damaged lungs repair themselves.
A Cellular Traffic Controller Gone Wrong
FUS is a protein that usually lives in the cell’s nucleus, where it helps manage how RNA—the working copy of our genes—is processed and used. In brain diseases such as ALS, FUS can misbehave, leaving the nucleus, clumping in the cell’s outer regions, and disturbing normal cell function. The authors asked whether a similar misbehavior might drive scarring in IPF. They studied lung fibroblasts—the connective tissue cells that lay down scar material—from patients with IPF and from healthy donors. In IPF cells, FUS levels were higher overall and, crucially, much more FUS was found in the cytoplasm than in healthy cells. Using high-resolution electron microscopy, they confirmed that this protein was abnormally abundant outside the nucleus, hinting that its normal control over RNA might be distorted in fibrotic lungs. 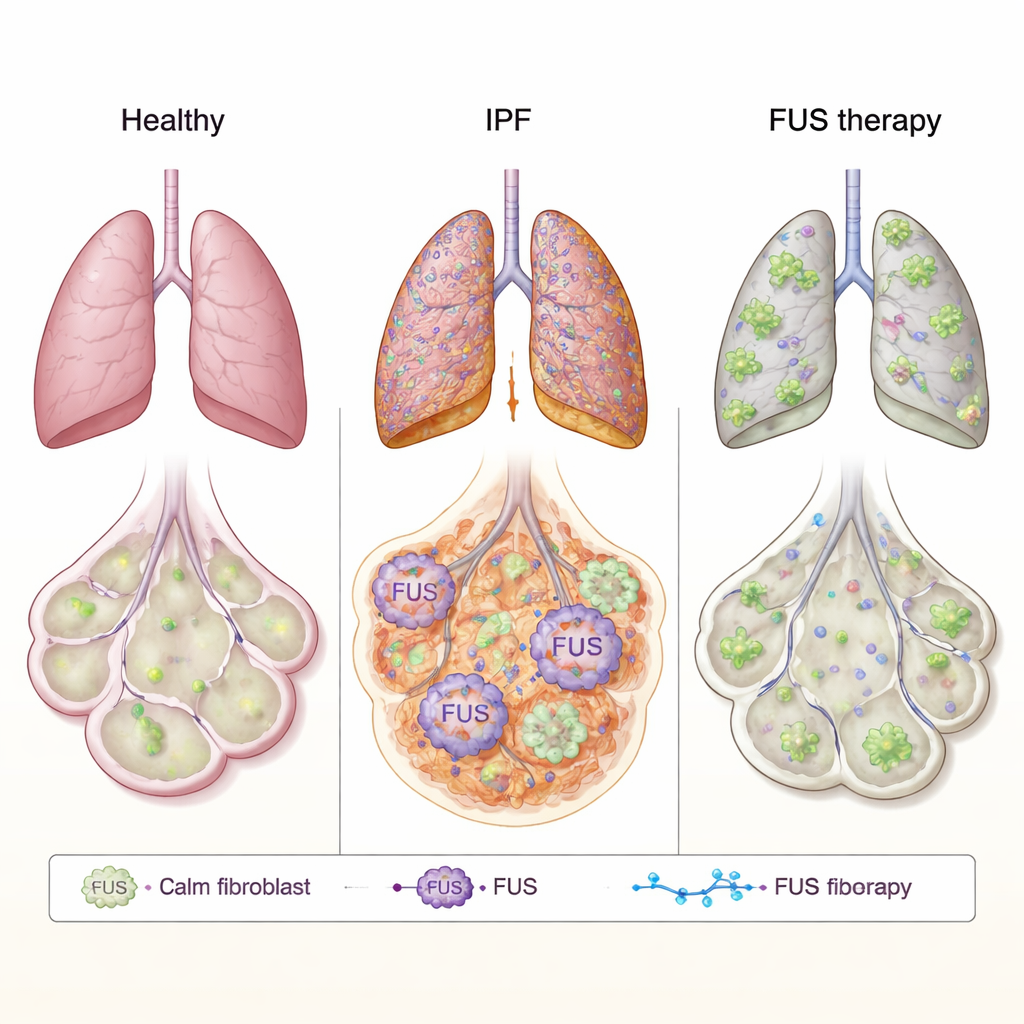
How FUS Fuels Scarring Cells
To see what this misbehaving protein actually does, the researchers boosted FUS in healthy fibroblasts and dialed it down in IPF fibroblasts. Extra FUS pushed healthy cells to divide more quickly, while reducing FUS in IPF cells slowed their growth and movement—two behaviors that are central to scar formation. The team then used a technique that “freezes” protein–RNA partnerships in place and reads out which RNAs are bound to FUS. In IPF fibroblasts, FUS was found glued to many genetic messages that promote fibrosis, including those that code for collagen, growth factors like TGF‑β, and inflammatory signals. In other words, FUS was acting as a hub connecting a whole network of pro‑scarring messages.
Silencing the Signal with a Precision Drug
The study tested an antisense oligonucleotide called ION363—a short, chemically modified strand designed to bind FUS RNA and trigger its destruction. When IPF fibroblasts were treated with ION363, FUS levels dropped, the cells slowed their proliferation and migration, and key scar‑building genes went quiet. Importantly, this effect did not rely on killing the cells or forcing them into old age; instead, it seemed to reset their behavior. When the same treatment was applied to thin slices of IPF lung tissue kept alive in the lab, large groups of genes linked to the extracellular matrix, inflammation, and abnormal epithelial lining were toned down, while genes associated with healthy surfactant production and alveolar function were boosted. The treatment also reduced collagen staining and increased markers of functional lung surface cells, suggesting a shift from scarring toward repair.
Helping Damaged Air Sacs Regrow
Because the tiny air‑sac–lining cells, known as type II alveolar cells, are crucial for lung repair, the researchers built three‑dimensional “alveolospheres” from patient cells to mimic miniature lung units. In cultures from IPF patients, these structures normally survive poorly. With ION363 treatment, more alveolospheres formed, grew larger, and showed greater lysosomal activity—a hallmark of active renewal. Detailed staining revealed more cells bearing markers of mature gas‑exchanging cells, indicating that silencing FUS not only calmed fibroblasts but also encouraged the injured epithelium to rebuild a healthier surface. 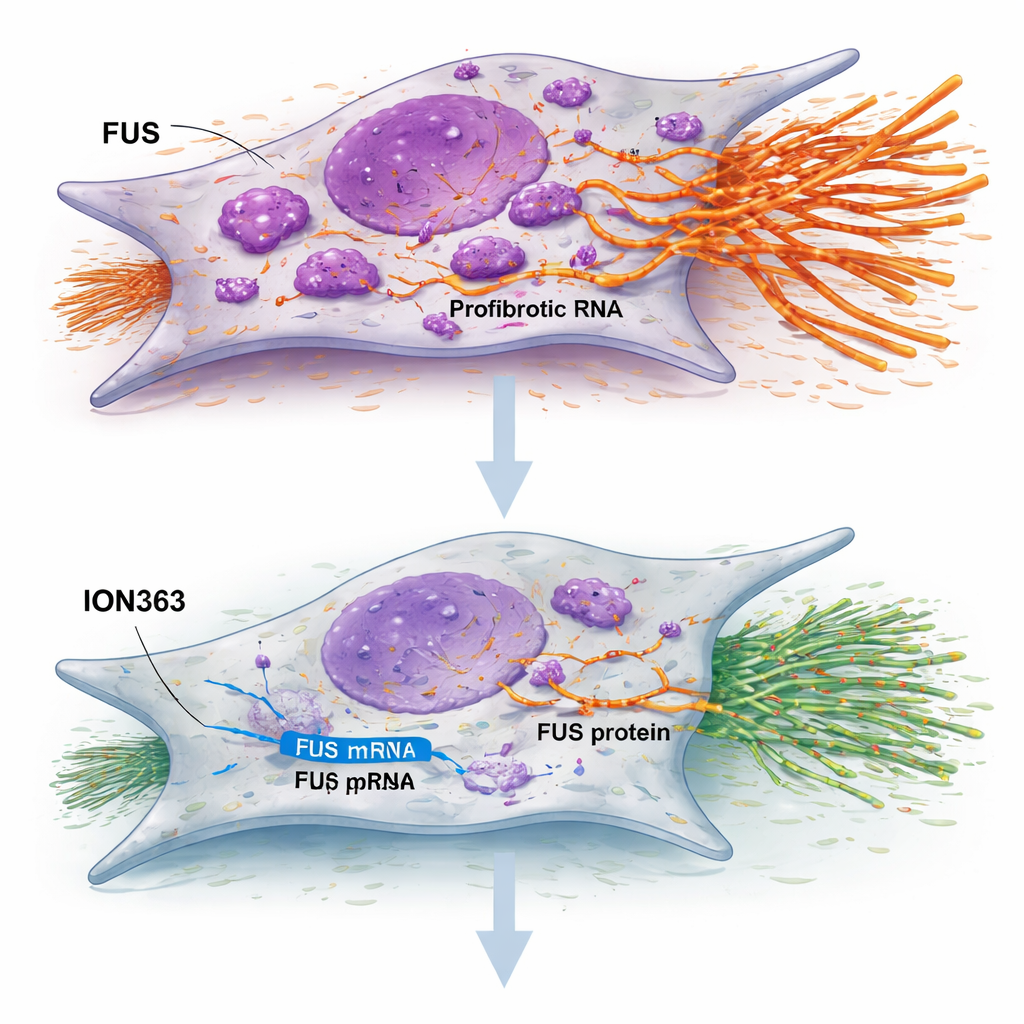
What This Could Mean for Patients
Taken together, the work paints FUS as a master switch in IPF that links overactive scar‑forming fibroblasts with failing repair of the delicate air sacs. By turning down FUS with a targeted antisense drug, the researchers could reduce profibrotic gene programs, ease collagen buildup, and promote regeneration in patient‑derived lung models. Although this approach is still at the laboratory stage and will require careful testing in animal models and clinical trials, it suggests that IPF might one day be treated not only by slowing scarring, but by directly rebalancing the cellular programs that control lung injury and repair.
Citation: Katariya, B.B., Chillappagari, S., Arnold, L. et al. Targeting fused in sarcoma (FUS): a novel antisense strategy for treating idiopathic pulmonary fibrosis. Sig Transduct Target Ther 11, 70 (2026). https://doi.org/10.1038/s41392-026-02585-9
Keywords: idiopathic pulmonary fibrosis, antisense oligonucleotide, FUS protein, lung fibrosis, alveolar repair