Clear Sky Science · de
Tumorspezifische HORMAD1‑Expression stört die Mitose‑Arretierung und erhöht die Empfindlichkeit gegenüber Mitosekinase‑Inhibitoren
Warum das für die Krebstherapie wichtig ist
Bei der Zellteilung verlassen sich Zellen auf komplexe Sicherheitskontrollen, um die richtige Chromosomenausstattung weiterzugeben. Krebs kapert oder schwächt diese Schutzmechanismen häufig, was zu chaotischen Genomen führt, die die Krankheit vorantreiben und zugleich das Ansprechen von Tumoren auf Medikamente prägen. Diese Studie zeigt, wie ein ungewöhnliches Protein namens HORMAD1, das normalerweise nur in Keimzellen aktiv ist, in vielen aggressiven Brustkrebserkrankungen und anderen Tumoren fehlaktiviert wird. Durch eine subtile Sabotage eines wichtigen Kontrollpunkts der Zellteilung macht HORMAD1 Krebszellen instabiler — aber zugleich ungewöhnlich verwundbar gegenüber einer neuen Klasse experimenteller Wirkstoffe.
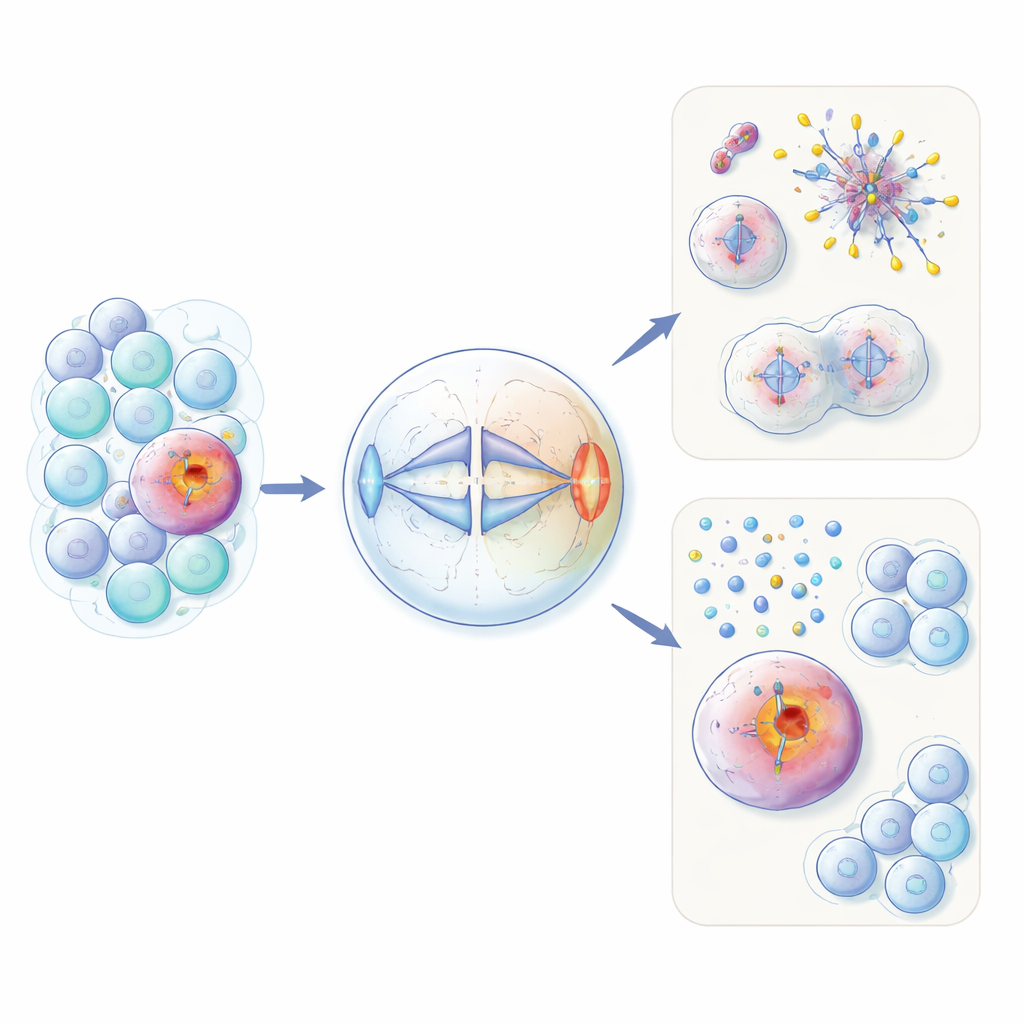
Ein fehlplatzierter Fruchtbarkeitsfaktor in Krebszellen
HORMAD1 tritt normalerweise nur in Keimzellen auf, wo Eizellen und Spermien entstehen. Dort hilft es bei der DNA‑Umordnung und Qualitätskontrolle während der speziellen Zellteilung Meiose. Die Autoren zeigen, dass dieses Protein in etwa 60 % der triple‑negativen Brustkrebse — sowie in Untergruppen mehrerer anderer Tumorarten — unpassend wieder aktiviert ist. Anhand sowohl gentechnisch veränderter nicht‑kanzeröser Zellen als auch Krebszelllinien fanden sie heraus, dass zusätzliches HORMAD1 die gleichmäßige Trennung der Chromosomen in der normalen Mitose stört. HORMAD1‑expressierende Zellen zeigten mehr nachhängende Chromosomen, zusätzliche oder fehlende Chromosomen (Aneuploidie) und kleine DNA‑haltige „Mikronuklei“, alles Kennzeichen genomischen Chaos, wie es in aggressiven Krebsen zu sehen ist.
Wie die Sicherheitschecks der Zellteilung normalerweise funktionieren
Damit die Teilung korrekt verläuft, bildet eine Zelle einen Spindelapparat aus Mikrotubuli, der an jedem Chromosom anhaftet. Ein Überwachungssystem, bekannt als Spindle‑Assembly‑Checkpoint, wirkt wie eine spannungsabhängige Bremse: Wenn ein Chromosom nicht richtig befestigt ist, blockiert die Bremse den Fortschritt und verhindert die Trennung, bis Fehler behoben sind. Mehrere Enzyme, die Mitosekinasen genannt werden, darunter MPS1, Aurora B und BUB1, helfen, fehlerhafte Anheftungen zu erkennen und die „Fehlerkorrektur“ zu fördern, damit jede Tochterzelle die richtige Chromosomenausstattung erhält. Die Störung dieses Systems kann sowohl die Krebsentstehung fördern als auch spezielle Verwundbarkeiten erzeugen, die bestimmte Medikamente ausnutzen können.
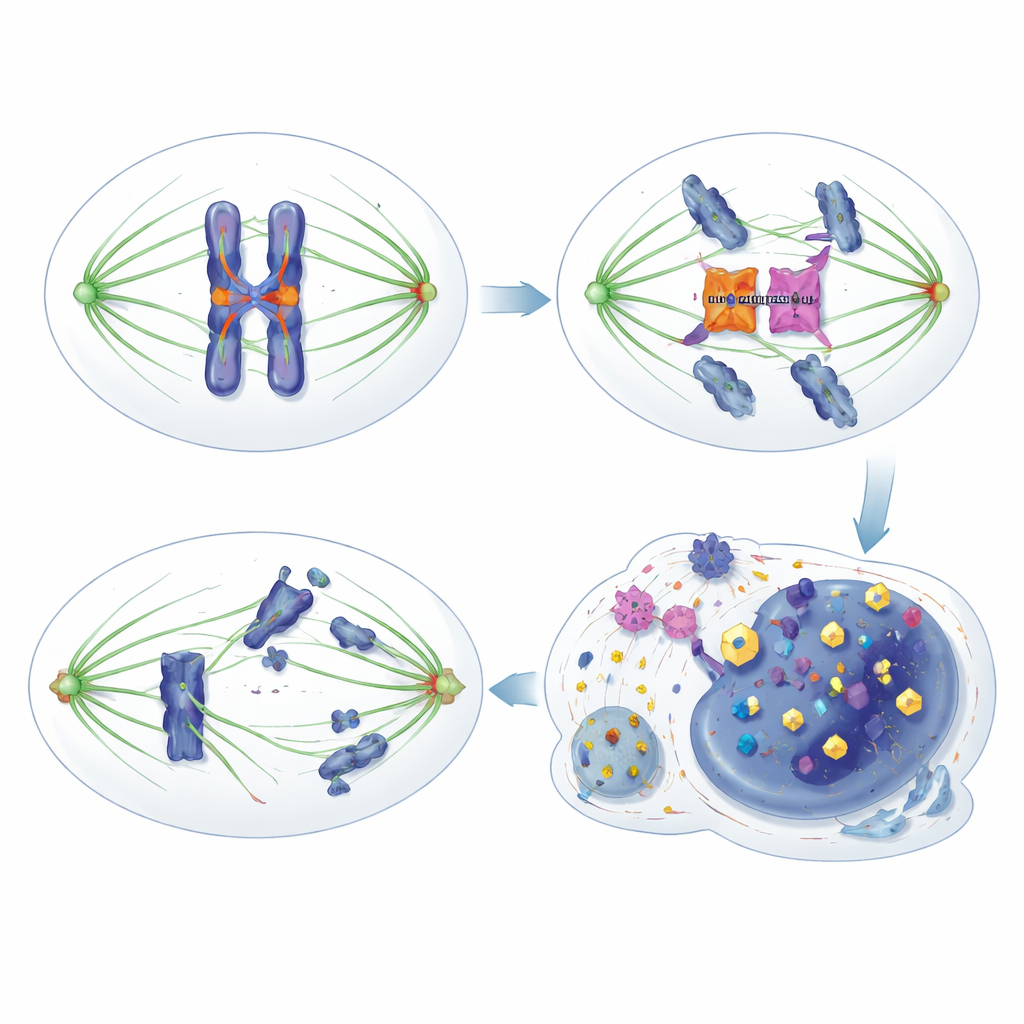
HORMAD1 schwächt die Bremse auf subtile Weise
Die Forscher stellten fest, dass HORMAD1 diese Sicherheitsbremse auf eine subtile, aber bedeutsame Weise untergräbt. Statt klassische Checkpoint‑Komponenten wie das HORMA‑Protein MAD2L1 direkt zu stören, bindet HORMAD1 unmittelbar an die Kinasen‑Komponente Aurora B. Aurora B arbeitet normalerweise mit einem Partnerprotein, INCENP, zusammen, um voll aktiv zu werden und Zielproteine an Zentromeren und Kinetochoren zu modifizieren — kritische Stellen an Chromosomen, an denen Spindelfasern ansetzen. Wenn HORMAD1 in teilenden Tumorzellen vorhanden ist, konkurriert es mit INCENP um die Bindung an Aurora B, schwächt deren Partnerschaft und dämpft dadurch die Aktivität von Aurora B. Infolgedessen werden die üblichen Phosphorylierungs‑Signale von Aurora B auf mehreren Targets abgeschwächt, die Fehlerkorrektur wird weniger effektiv, und der Checkpoint wird „undicht“: Zellen verlassen die Mitose zu früh, selbst wenn Anheftungen fehlerhaft sind, wodurch es zu fehlverteilten Chromosomen und genomischer Instabilität kommt.
Vom Schwachpunkt zur therapeutischen Chance
Weil HORMAD1 nur teilweise Aurora B und verwandte Schutzmechanismen schwächt, bleiben Krebszellen gerade noch lebensfähig, sind aber stark von der verbliebenen Funktion der Mitosekinasen abhängig, um wiederholte fehlerhafte Teilungen zu überstehen. Das Team prüfte dies, indem es HORMAD1‑positive und HORMAD1‑negative Zellen experimentellen Inhibitoren von MPS1, Aurora B und BUB1 aussetzte. In mehreren Modellen machte HORMAD1‑Expression die Zellen deutlich empfindlicher gegenüber diesen Wirkstoffen, wodurch ihre Fähigkeit zu proliferieren oder Kolonien zu bilden drastisch reduziert wurde. Die genetische Depletion von BUB1 war besonders tödlich, und zwar nur in Anwesenheit von HORMAD1, was eine starke, selektive Abhängigkeit offenbarte. In Mausmodellen mit patientenabgeleiteten triple‑negativen Brusttumoren schrumpften oder wuchsen Tumoren mit hohem HORMAD1‑Spiegel unter Behandlung mit einer nanopartikelbasierten Formulierung eines Aurora‑B‑Inhibitors langsamer, während HORMAD1‑negative Tumoren derselben Behandlung weitgehend widerstanden.
Was das für Patientinnen und Patienten bedeutet
Für Außenstehende wirkt HORMAD1 in Krebszellen wie ein zweischneidiges Schwert: Es treibt Tumorzellen zu größerer chromosomaler Unordnung, was die Krankheit fördern kann, macht sie dabei aber zugleich abhängig von wenigen verbliebenen Zellteilungs‑Schutzmechanismen. Die Studie zeigt, dass dieses fehlplatzierte Fruchtbarkeitsprotein einen Schlüsselcheckpoint schwächt, indem es Aurora B fehlleitet, und HORMAD1‑positive Tumoren damit besonders anfällig für Wirkstoffe sind, die Aurora B, MPS1 oder BUB1 angreifen. Da HORMAD1 in normalen Geweben weitgehend fehlt, aber in einer klaren Untergruppe von Tumoren vorkommt, könnte es als Biomarker dienen, um Patientinnen und Patienten zu identifizieren, die am ehesten von diesen aufkommenden Mitosekinase‑Inhibitoren profitieren — und damit neue, gezielte Therapieoptionen für schwer zu behandelnde Krebsarten wie den triple‑negativen Brustkrebs eröffnen.
Zitation: Walker, C., Kollarovic, G., Weekes, D. et al. Tumour specific HORMAD1 expression perturbs mitotic arrest and drives sensitivity to mitotic kinase inhibitors. Nat Commun 17, 2157 (2026). https://doi.org/10.1038/s41467-026-69561-3
Schlüsselwörter: HORMAD1, triple-negativer Brustkrebs, chromosomale Instabilität, Aurora‑B‑Kinasen, Mitose‑Checkpoint‑Inhibitoren