Clear Sky Science · de
Die Hemmung von HSP27 aktiviert das Zusammenspiel von XBP1s und CerS1, das DRP1‑getriebene Mitophagie auslöst, wodurch Zelltod verhindert und der lytische Zyklus von KSHV in Zellen des Primary Effusion Lymphoms gefördert wird
Wenn Zellstress zur zweischneidigen Waffe wird
Unsere Zellen überstehen tägliche Schädigungen, indem sie Notfallprogramme hochfahren, die Schäden reparieren und das Überleben sichern. Krebszellen können diese Programme jedoch kapern, um zu wachsen und darin ruhende Viren zu schützen. Dieser Artikel untersucht, wie die Blockade eines einzigen stressschützenden Proteins in einem seltenen Lymphom die Tumorzellen nicht nur in Richtung Zelltod drängt, sondern zugleich einem darin verborgenen Virus ein Zeitfenster eröffnet, um sich zu reaktivieren und zu vermehren. Das Verständnis dieses empfindlichen Gleichgewichts könnte helfen, Therapien zu entwickeln, die Krebszellen töten, ohne dem Virus eine Ausbreitung zu ermöglichen.
Ein verborgenes Virus in einem aggressiven Lymphom
Primary Effusion Lymphom ist ein hochaggressiver Krebs der B‑Zellen, einer Art weißer Blutkörperchen. Die meisten dieser Tumorzellen tragen einen ruhenden Mitbewohner: das Kaposi‑Sarkom‑assoziierte Herpesvirus (KSHV). Im latenten Zustand produziert das Virus nur wenige Proteine und versteckt sich im Wirtsgenom. Bestimmte Stressfaktoren können es in eine aktive, lytische Phase treiben, in der es sich kopiert und neue Partikel produziert, was üblicherweise die Wirtszelle tötet. Die Tumorzellen selbst sind auf mehrere Stressantwortsysteme angewiesen, darunter sogenannte Hitzeschockproteine und die Antwort auf falsch gefaltete Proteine (unfolded protein response), die ihnen helfen, mit fehlgefalteten Proteinen, gestörter Lipidstoffwechsel und Schäden an ihren energieerzeugenden Mitochondrien fertigzuwerden. 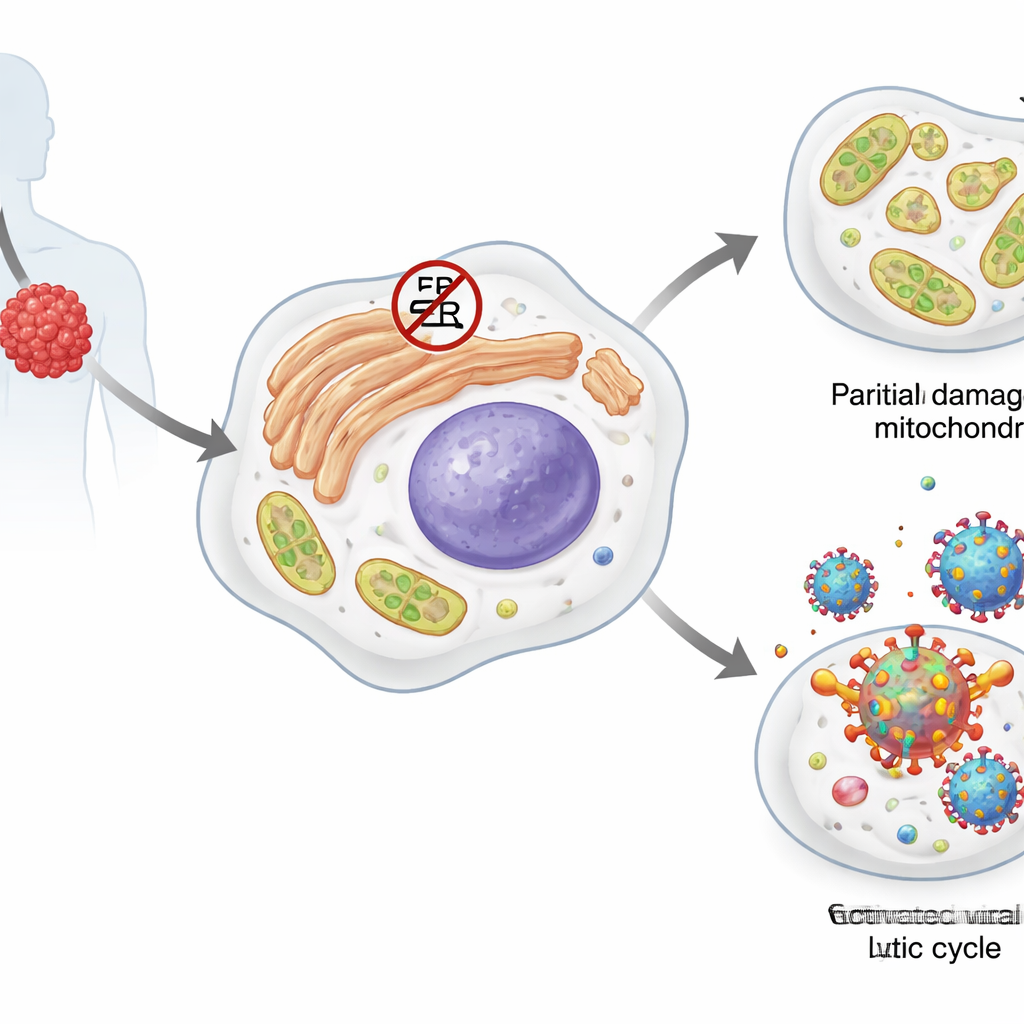
Blockade eines zellulären Leibwächters
Die Forschenden konzentrierten sich auf HSP27, ein kleines Hitzeschockprotein, das Zellen vor Stress schützt. Mit einem chemischen Inhibitor namens J2 oder durch Stilllegung des Gens mit kleinen RNAs reduzierten sie die HSP27‑Aktivität in im Labor kultivierten Lymphomzellen. Dadurch überlebten die Zellen weniger gut, und in einem internen Membrannetzwerk, dem endoplasmatischen Retikulum, löste sich ein starker Stresssignalweg aus. Marker dieser Antwort, einschließlich schützender und pro‑apoptotischer Faktoren, stiegen an, und ein wichtiger Schalter namens XBP1s wurde aktiviert. Gleichzeitig zeigten die Zellen verstärkte Anzeichen für programmierten Zelltod, was bestätigt, dass das Entfernen von HSP27 sie an einen Kipppunkt zwischen Überleben und Untergang bringt.
Eine Stressschleife, die mit Lipiden kommuniziert
Stress im endoplasmatischen Retikulum ist eng mit der Lipidverarbeitung der Zelle verknüpft. Das Team stellte fest, dass die Hemmung von HSP27 die Spiegel von CerS1 erhöhte, einem Enzym, das ein bestimmtes Lipidmolekül namens C18‑Ceramid herstellt. Als sie XBP1s chemisch blockierten, verschwand der Anstieg von CerS1, was zeigt, dass XBP1s unter diesen Bedingungen das CerS1‑Gen mit aktiviert. Bemerkenswerterweise senkte die Hemmung von CerS1 wiederum XBP1s, was eine positive Rückkopplungsschleife offenbart: Jeder Faktor stützt den anderen. Dieser molekulare Austausch verändert nicht nur den Lipidstoffwechsel, sondern stärkt auch die Fähigkeit der Zelle, sich an ER‑Stress anzupassen, selbst während pro‑tödliche Signale aufgebaut werden.
Mitochondrien recycelt statt zerstört
Stress in einem Zellkompartiment wirkt oft auf die Mitochondrien, die kleinen Kraftwerke der Zelle, über. Nach der Blockade von HSP27 produzierten die Lymphomzellen mehr reaktive Sauerstoffspezies, ein Hinweis auf mitochondriale Störungen, und zeigten erhöhte Werte von DRP1, einem Protein, das Mitochondrien in kleinere Stücke teilt. Die Autorinnen und Autoren zeigten, dass die XBP1s–CerS1‑Schleife für die Erhöhung von DRP1 verantwortlich war. Dies wiederum löste Mitophagie aus, einen Qualitätskontrollprozess, bei dem beschädigte Mitochondrien in Membranen verpackt und zu zellulären "Recyclingzentren", den Lysosomen, transportiert werden. Mit fluoreszierenden Farbstoffen und Proteinmarkern bestätigten sie, dass Mitochondrien selektiv entfernt wurden. Als sie DRP1 chemisch oder genetisch blockierten, nahm diese Mitophagie ab und die Zellen starben leichter, was bedeutet, dass das mitochondriale Recycling den gestressten Tumorzellen tatsächlich half, durchzuhalten. 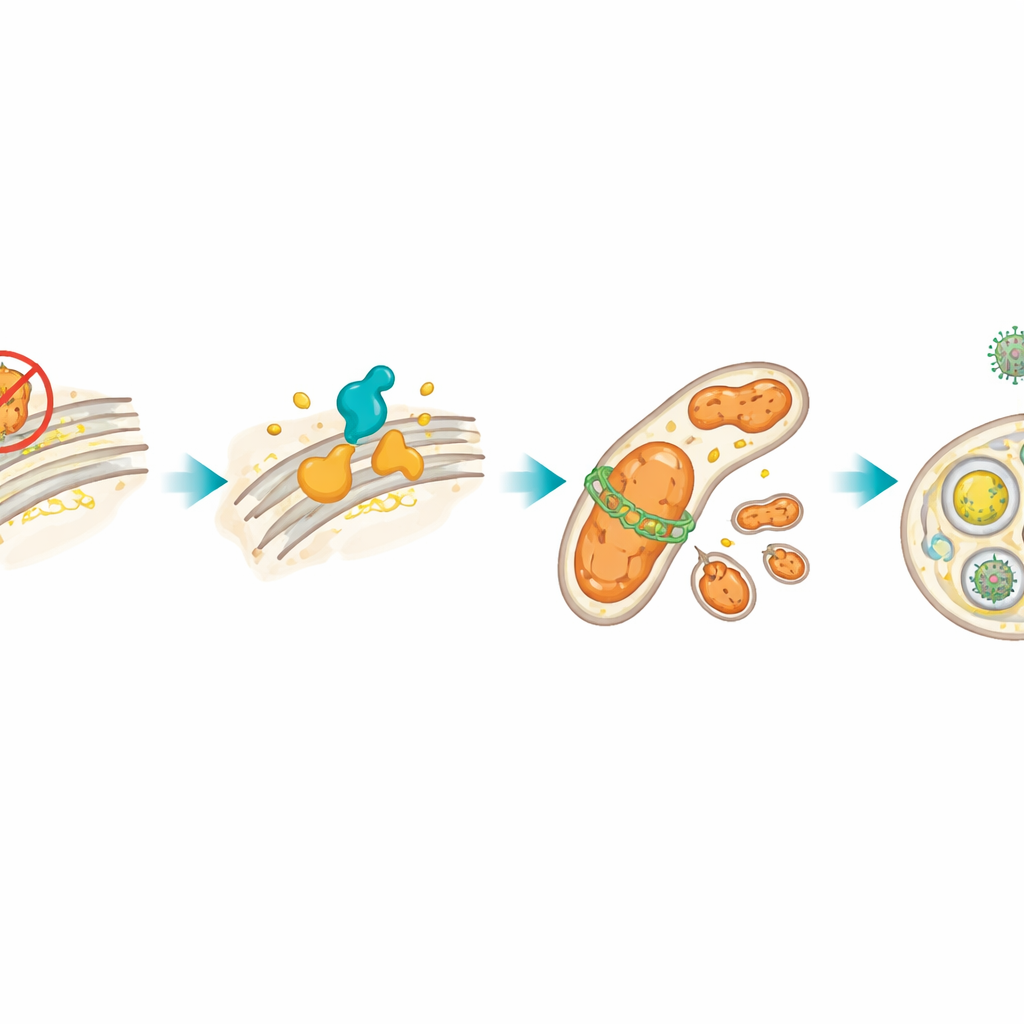
Dem Virus Zeit zur Flucht geben
Die gleiche Mitophagie, die die Tumorzellen schützte, kam auch KSHV zugute. Die Aktivierung von XBP1s, die Anreicherung von C18‑Ceramid und verstärkte mitochondriale Fragmentierung werden alle mit der Reaktivierung dieses Virus in Verbindung gebracht. Hier führten die Hemmung von HSP27 dazu, dass mehr Zellen frühe und späte virale Proteine exprimierten — deutliche Zeichen lytischer Replikation. Die Blockade von DRP1 und damit der Mitophagie verringerte diese Virusreaktivierung. Die Autorinnen und Autoren schlagen vor, dass Mitophagie, indem sie das Überleben der Zelle unter Stress leicht verlängert, KSHV Zeit gibt, seinen Replikationszyklus zu vollenden und die sterbende Zelle zu verlassen, wodurch neue Ziele infiziert werden und zur Krebsentstehung beitragen könnten.
Was das für künftige Therapien bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft, dass HSP27 als zentrale Schaltstelle fungiert, die steuert, wie Lymphomzellen mit Stress umgehen, wie sie beschädigte Mitochondrien recyceln und wann ein krebsassoziiertes Virus erwacht. Das Ausschalten von HSP27 löst eine Kaskade aus, die einerseits das Zellüberleben untergräbt und andererseits paradoxerweise die Zellen vorübergehend durch Mitophagie schützt, während KSHV replizieren kann. Therapeutisch könnte die Kombination von HSP27‑Hemmung mit Wirkstoffen, die die DRP1‑getriebene Mitophagie blockieren, die Tumorzellen schneller zum Tod führen und gleichzeitig die Chance des Virus zur Ausbreitung verringern — eine zweigleisige Strategie gegen dieses tödliche Lymphom.
Zitation: Gonnella, R., Corrado, V., Scaffidi, G.F. et al. Inhibiting HSP27 activates the XBP1s/CerS1 interplay, which triggers DRP1-driven mitophagy, thereby protecting against cell death and promoting the KSHV lytic cycle in primary effusion lymphoma cells. Cell Death Discov. 12, 118 (2026). https://doi.org/10.1038/s41420-026-02979-2
Schlüsselwörter: primary effusion lymphoma, Kaposi‑Sarkom‑assoziiertes Herpesvirus, zelluläre Stressantwort, Mitophagie, Hitzeschockprotein HSP27