Clear Sky Science · de
Ein lebensfähiges kinase-inaktives RIPK3 D143N-Mausmodell zeigt seine Gerüstfunktion bei TNF‑induzierten Entzündungsstörungen
Warum diese Mausstudie für Entzündungen wichtig ist
Viele schwere Erkrankungen — von tödlichen Infektionen bis zu Autoimmun‑Schüben — werden nicht nur durch Erreger oder Gene angetrieben, sondern durch eine fehlregulierte, sich selbst verstärkende Entzündung. Ein Protein namens RIPK3 galt lange als Schlüsselfigur bei einer gewalttätigen Form des Zelltods, die solche Entzündungen antreibt, und erschien deswegen als attraktives Ziel für Medikamente. RIPK3 hat jedoch auch andere, weniger verstandene zelluläre Rollen. Diese Studie beschreibt eine neue Mauslinie, die die tödliche Aktivität von RIPK3 sauber von seiner signalgebenden "Gerüst"-Funktion trennt, zeigt, wie beide zur Entzündung beitragen, und weist auf neue therapeutische Ansätze hin.
Zwei Arten, wie ein Todesprotein wirken kann
Zellen können auf geordnete oder chaotische Weise sterben. Beim stillen, "leisen" Zelltod recycelt der Körper Zellbestandteile ohne große Alarmbereitschaft. Bei einer unordentlicheren Form, der Nekroptose, platzen Zellen auf und setzen Innenstoffe frei, was starke Immunreaktionen auslöst. RIPK3 steht im Mittelpunkt der Nekroptose: Sobald es aktiviert ist, schaltet es ein anderes Protein ein, das Löcher in die Zellmembran macht. Frühere Arbeiten deuteten jedoch an, dass RIPK3 auch klassische, caspase‑getriebene Apoptose fördern und entzündliche Signalwege ankurbeln kann, ohne Zellen zu töten. Diese Rollen auseinanderzuhalten war schwer, weil bisherige inaktive RIPK3‑Varianten entweder Embryonen töten oder die Proteinmenge stark verringern und so das Studium seiner normalen Gerüstfunktion verhindern.
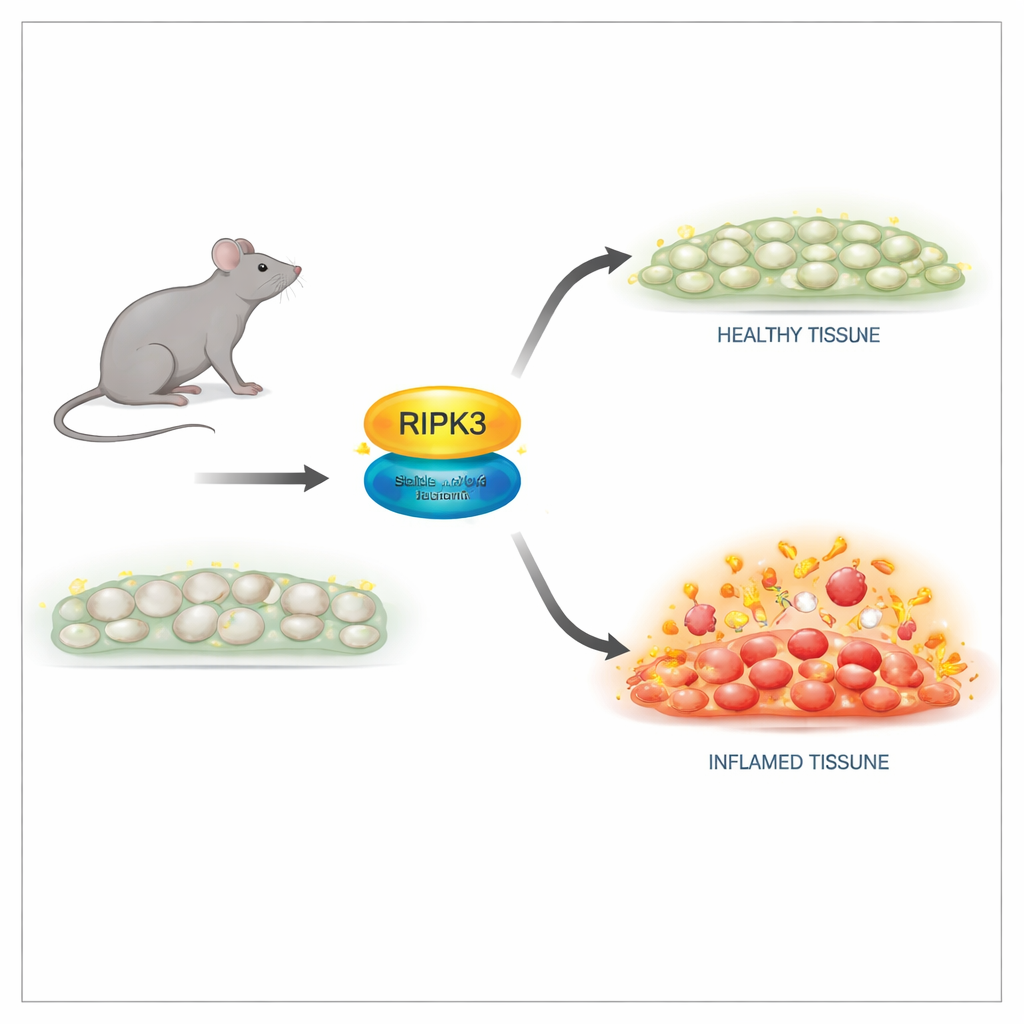
Ein sichererer Weg, die Todesfunktion abzuschalten
Die Forscher erzeugten Mäuse mit einer dezenten Veränderung an einer einzigen Position des RIPK3‑Proteins, bezeichnet D143N, die seine Enzymaktivität abschaltet, aber die Struktur bewahrt. In Zellen dieser Mäuse waren RIPK3‑Proteinspiegel und Gewebearchitektur normal, und die Tiere wurden gesund geboren und entwickelten sich wie ihre gesunden Wurfgeschwister. Wichtig ist, dass Zellen mit der D143N‑Version vollständig gegen multiple Nekroptose‑Auslöser resistent waren, darunter Signale von Tumornekrosefaktor (TNF), Toll‑like‑Rezeptoren und virale Infektionen. Das mutierte RIPK3 konnte seinen downstream‑Partner nicht mehr aktivieren oder den zerstörerischen Komplex zur Membranruptur bilden, löste aber keine spontane Apoptose aus und vermied damit die tödlichen Nebenwirkungen früherer RIPK3‑Mutanten.
Entwicklung von Krankheit trennen
Eine der bekanntesten Rollen von RIPK3 zeigt sich in Embryonen, denen ein weiteres Schlüsselprotein, Caspase‑8, fehlt: Ohne Caspase‑8 tötet RIPK3‑vermittelte Nekroptose den Embryo. In dieser Studie rettete die Einführung der D143N‑Version von RIPK3 diese sonst nicht lebensfähigen Mäuse vollständig. Sie entwickelten sich normal und waren fruchtbar, was beweist, dass die tödliche Aktivität von RIPK3 für die normale Entwicklung entbehrlich ist, sofern seine Struktur erhalten bleibt. Bei erwachsenen Mäusen, die jedoch mit hoher TNF‑Dosis einem schockähnlichen Entzündungssyndrom ausgesetzt wurden, änderte sich das Bild. Tiere, die RIPK3 vollständig fehlten, waren stark geschützt vor Tod, Gewebeschäden und entzündlichen Molekülen im Blut. Mäuse mit der D143N‑Variante, obwohl ohne Nekroptose, waren nur teilweise geschützt. Das deutet darauf hin, dass RIPK3s nicht‑tödliche Gerüstfunktion weiterhin zur Entzündung beiträgt.
Gerüst‑Signale, die das Feuer anfachen
Um diesen nicht‑letalien Beitrag zu verstehen, untersuchte das Team die Genaktivität im Darm von TNF‑behandelten Mäusen. Bei RIPK3‑defizienten Tieren waren viele entzündliche Gene stark gedämpft. In D143N‑Mäusen war die Unterdrückung hingegen schwächer, und Gene, die mit Interferon‑ und angeborenen Immunantworten verknüpft sind, blieben aktiver. Auf Proteinebene aktivierte TNF robust die JAK–STAT1‑ und ERK‑Signalwege in normalen und D143N‑Mäusen, während diese Aktivierung nahezu vollständig fehlte, wenn RIPK3 ganz gelöscht war. Das zeigte, dass selbst ohne seine Todesfunktion die physische Präsenz von RIPK3 in Signalkomplexen hilft, TNF‑Signale über JAK–STAT1 in ein pro‑entzündliches Programm zu übersetzen.
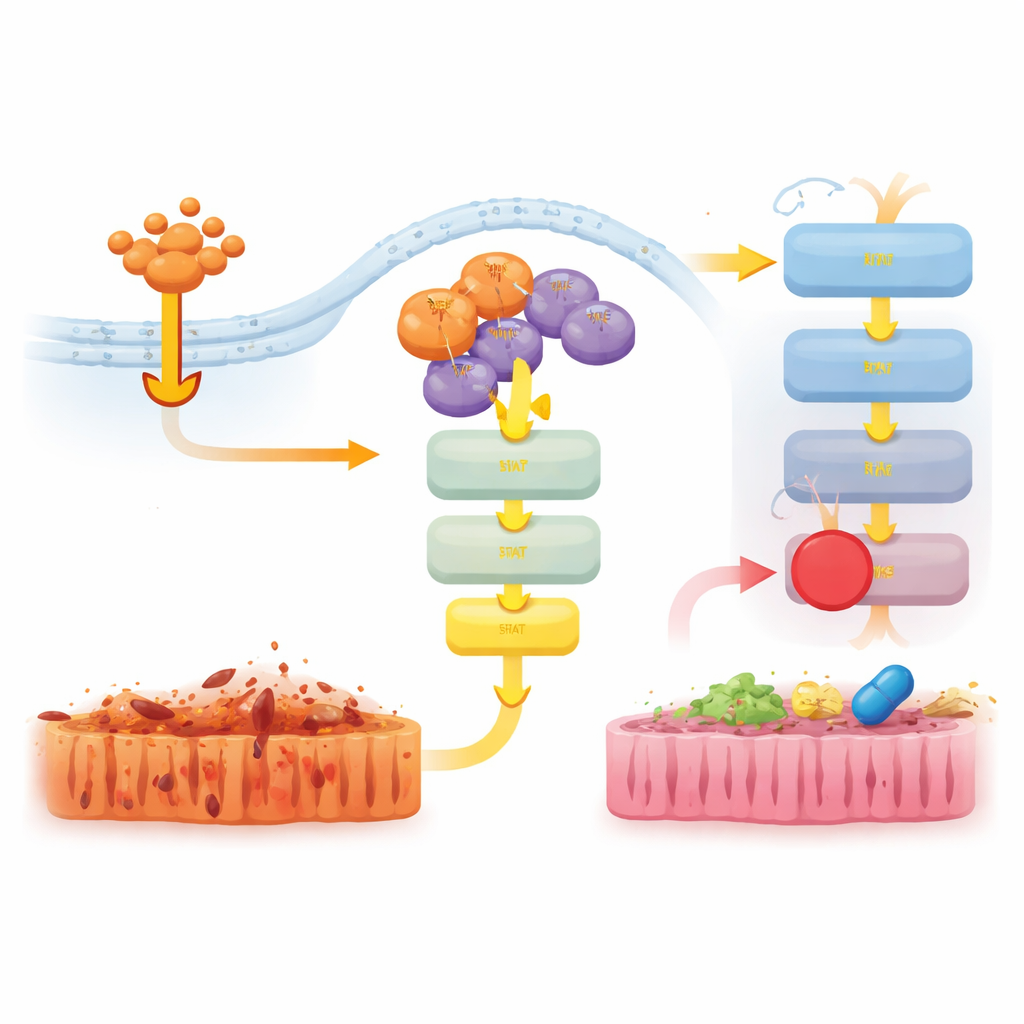
Schädliche Signale mit gezielten Medikamenten dämpfen
Die Forscher testeten dann, ob das Blockieren dieser downstream‑Wege die Erkrankung in D143N‑Mäusen während eines TNF‑induzierten Schocks lindern könnte. Die Behandlung der Tiere mit einem JAK1/2‑Inhibitor, nicht jedoch mit einem ERK‑Inhibitor, verringerte den Verlust der Körpertemperatur, senkte die Spiegel des Entzündungsmoleküls IL‑6 und milderte Darmgewebeschäden sowie Zelltod. Ein separater Inhibitor, der ein anderes Protein, RIPK1, blockiert, schützte die Mäuse ebenfalls stark und dämpfte die Aktivierung von JAK–STAT1 und ERK. Zusammengenommen deuten diese Ergebnisse darauf hin, dass RIPK3s Gerüstfunktion mit RIPK1 zusammenarbeitet, um JAK–STAT1 zu aktivieren und Entzündungen zu fördern, und dass das Unterbrechen dieser Signalwege Gewebeschäden reduzieren kann, selbst wenn die Nekroptose bereits blockiert ist.
Was das für künftige Therapien bedeutet
Über Jahre wurde RIPK3 hauptsächlich als Schalter für eine toxische Form des Zelltods betrachtet, und die Medikamentenentwicklung konzentrierte sich auf das Abschalten seiner Enzymaktivität. Diese Studie zeigt, dass das allein möglicherweise nicht ausreicht: RIPK3 kann weiterhin als physische Plattform fungieren, die entzündliche Signale über JAK–STAT1 verstärkt und so zu Schock und Gewebeschädigung beiträgt. Das neue D143N‑Mausmodell macht diese doppelten Rollen ungewöhnlich deutlich und stellt ein mächtiges Werkzeug bereit, um zu untersuchen, wann und wie jede Funktion in verschiedenen Erkrankungen relevant ist. Für Patientinnen und Patienten legt die Arbeit nahe, dass eine Kombination aus RIPK3‑ oder RIPK1‑gerichteten Wirkstoffen mit JAK–STAT1‑Blockern die schädliche Entzündung bei TNF‑ und verwandten Zytokin‑getriebenen Erkrankungen effektiver dämpfen könnte.
Zitation: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
Schlüsselwörter: RIPK3, Nekroptose, Entzündung, TNF-Schock, JAK-STAT1