Clear Sky Science · de
Kurzketten-Acyl‑CoA‑Dehydrogenase initiiert mtDNA‑Demethylierung und Leckage zur Stärkung der antitumoralen Immunität beim kolorektalen Krebs
Warum unsere eigenen Zellen manchmal Krebs vor dem Immunsystem verbergen
Kolorektales Karzinom gehört zu den tödlichsten Tumoren weltweit, unter anderem weil die Immunabwehr des Körpers ihn oft nicht erkennt oder angreift. Diese Studie zeigt eine unerwartete Verbindung zwischen der Fettverbrennung von Krebszellen, der Art und Weise, wie ihre „Kraftwerke“ (Mitochondrien) mit ihrer DNA umgehen, und der Frage, ob das Immunsystem auf den Tumor aufmerksam wird. Indem die Forschenden diese Ereigniskette nachvollziehen, rücken sie außerdem eine bekannte natürliche Verbindung, Hypericin, als möglichen Weg in den Fokus, die Immunantwort gegen kolorektalen Krebs wiederzubeleben.
Ein fehlender mitochondrialer „Wächter“ in Darmtumoren
Das Team begann mit der Analyse großer menschlicher und muriner Datensätze, um Stoffwechselgene zu finden, die bei kolorektalem Krebs beständig verändert sind. Ein Enzym stach hervor: die Kurzketten‑Acyl‑CoA‑Dehydrogenase, kurz ACADS, die normalerweise den Abbau kurzkettiger Fettsäuren in Mitochondrien unterstützt. In Patientenproben und mehreren Mausmodellen war ACADS in Tumorgewebe deutlich geringer ausgeprägt als im benachbarten gesunden Kolon. Reduzierten die Forschenden ACADS in mausbasierten Darmkrebszellen, wuchsen die Tumoren schneller und aggressiver; eine Erhöhung von ACADS verlangsamte das Tumorwachstum. Mäuse, denen ACADS spezifisch im Darmepithel fehlte, entwickelten in einem chemischen Modell des kolitisassoziierten Karzinoms mehr und größere Tumoren, was die Vorstellung stützt, dass ACADS im Darm eine tumorunterdrückende Rolle spielt.
Wie Tumoren die Alarmzeichen des Immunsystems dämpfen
Diese Wachstumsunterschiede ließen sich nicht allein durch veränderte Teilungsraten der Krebszellen in der Kultur erklären, die sich kaum änderten. Vielmehr förderte der Verlust von ACADS Tumorwachstum nur in Tieren mit intaktem Immunsystem, was auf Veränderungen im Tumormikromilieu hindeutet. Einzelzellanalysen menschlicher Kolorektaltumoren zeigten, dass Tumoren mit niedrigem ACADS von mehr Tumorzellen und immunsuppressiven Zellen umgeben waren — etwa myeloid‑abhängige suppressive Zellen, bestimmte Makrophagen und regulatorische T‑Zellen — und weniger hilfreichen T‑Zellen und natürlichen Killerzellen. Dieses Muster deutet auf eine „immununterdrückende Nachbarschaft“, die den Krebs schützt. 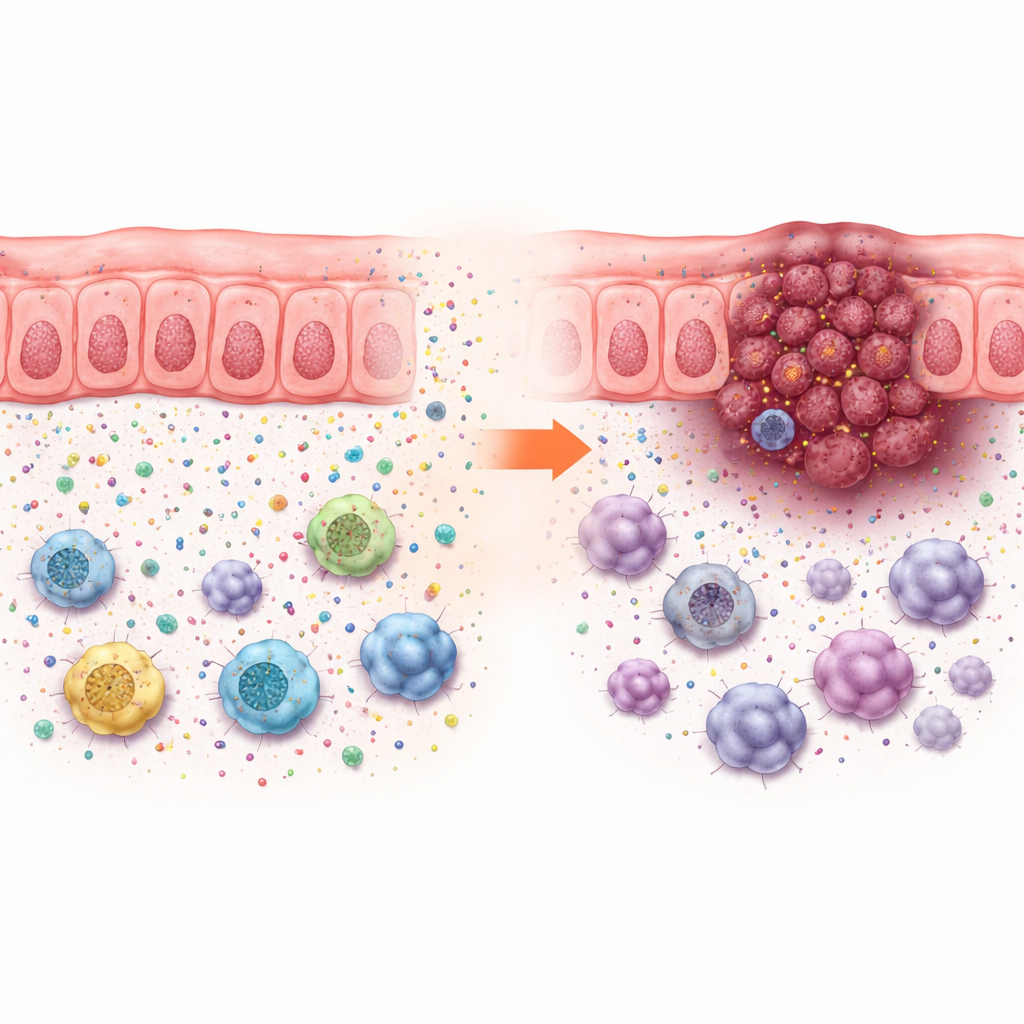
Mitochondriale DNA‑Leckage als verborgener Auslöser
Was verbindet ein fettsäureabbauendes Enzym mit einem Immun‑DNA‑Sensor? Die Antwort liegt in der mitochondrialen DNA (mtDNA). Unter Stress können Fragmente der mtDNA aus den Mitochondrien in die zelluläre Umgebung austreten, wo cGAS sie als Gefahrensignal erkennt. Die Forschenden zeigten, dass ACADS‑defiziente Krebszellen in diesem Kompartiment weniger mtDNA enthielten, obwohl die Gesamtmenge an mtDNA unverändert war. Das Blockieren der mtDNA‑Leckage in ACADS‑reichen Zellen schaltete cGAS–STING aus, womit bestätigt wurde, dass diese entwichenen DNA‑Fragmente der kritische Alarmgeber sind. Überraschenderweise konnten klassische Faktoren mitochondrialen Stresses wie reaktive Sauerstoffspezies, Calciumspitzen und größere Veränderungen der Mitochondrienform die Differenz nicht vollständig erklären. Stattdessen deuten die Ergebnisse auf die „Tore“ in der mitochondrialen Membran und vor allem auf chemische Markierungen der mtDNA selbst hin. 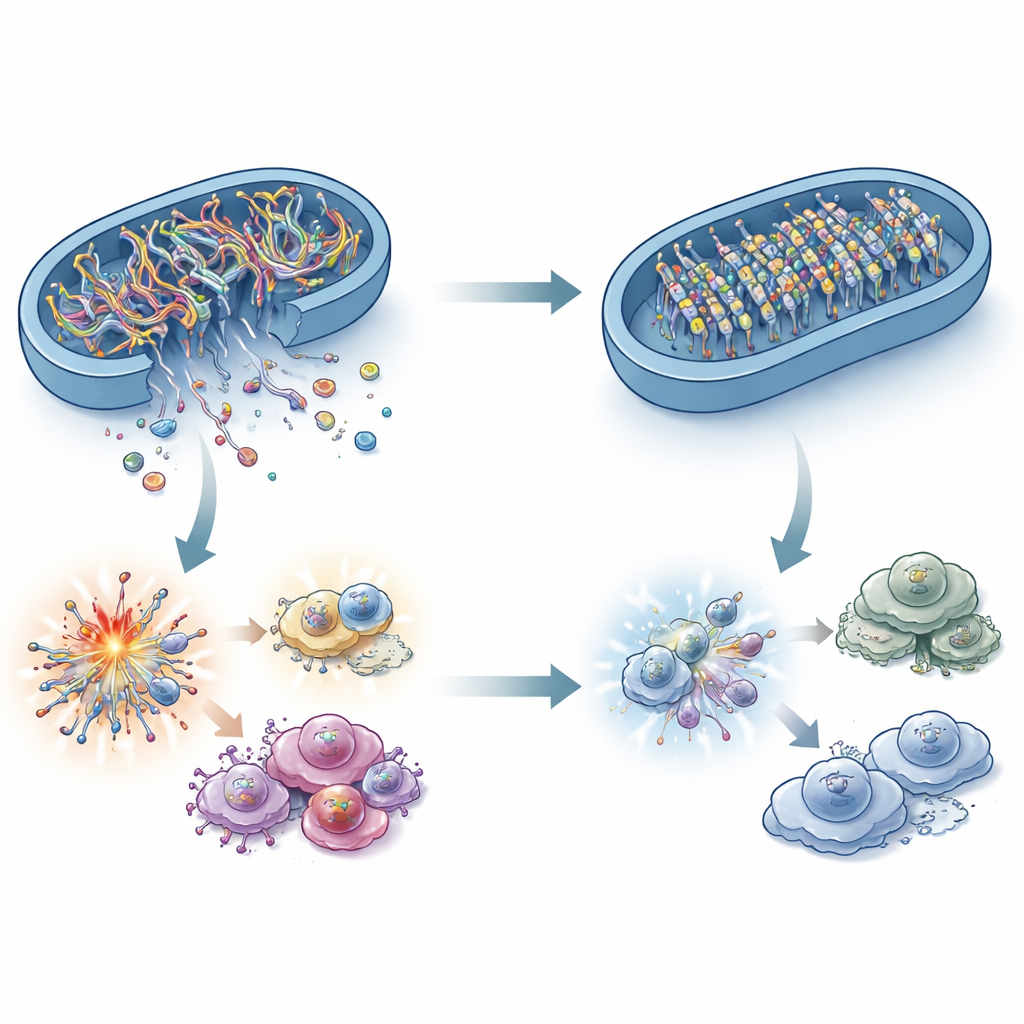
Ein DNA‑methylierender Partner, der den Alarm einschließt
Durch Proteingegenstücksanalysen zeigte sich, dass ACADS mit einer mitochondriellen Form des DNA‑Methylierungsenzymes DNMT1 assoziiert. Bei Verlust von ACADS häufte sich dieses mitochondriale DNMT1 an und brachte zusätzliche Methylgruppen auf die mtDNA. Diese Markierungen stabilisieren mtDNA und machen sie weniger bruch‑ und ausbruchanfällig. Eine Überexpression mitochondrialen DNMT1 reduzierte das Entweichen von mtDNA, dämpfte cGAS–STING‑Signale und beschleunigte das Tumorwachstum, während die Hemmung von DNMT1 mit dem Medikament Decitabin die mtDNA‑Leckage wiederherstellte und ACADS‑defiziente Tumoren verlangsamte. Patientenproben spiegelten diese Befunde wider: Niedriger ACADS‑Spiegel korrelierte mit hohem mitochondrialem DNMT1, schwächerer STING‑Signalgebung, weniger Effektorzellen vom T‑Zell‑Typ, mehr immunsuppressiven Zellen und einer schlechteren vorhergesagten Reaktion auf Checkpoint‑Immuntherapien.
Die Immunabwehr mit einer alten Verbindung wiedererwecken
Um zu prüfen, ob dieser Weg therapeutisch nutzbar ist, suchten die Forschenden rechnergestützt nach Molekülen, die an ACADS binden. Sie identifizierten Hypericin, ein natürliches Pigment, das zuvor als lichtaktivierbare Behandlung für bestimmte Hautlymphome getestet wurde. In kolorektalen Krebszellen erhöhte Hypericin die ACADS‑Spiegel, verringerte mitochondriales DNMT1, förderte die mtDNA‑Leckage und reaktivierte cGAS–STING‑Signale — Änderungen, die von der Anwesenheit von ACADS abhingen. In Maus‑Tumormodellen und in Kurzzeitkulturen menschlicher Kolorektaltumoren verkleinerte Hypericin die Tumoren oder verschob die Immunzellzusammensetzung hin zu einem aktiveren, T‑Zell‑reichen Zustand. Obwohl vor einer klinischen Anwendung weitere Arbeit nötig ist, deuten diese Ergebnisse darauf hin, dass das pharmakologische „Wiederanschalten“ von ACADS helfen könnte, einen kalten, immununterdrückten Tumor in einen empfindlicheren gegen Immuntherapie zu verwandeln.
Was das für Patientinnen und Patienten und zukünftige Behandlungen bedeutet
Einfach ausgedrückt zeigt diese Arbeit, dass sich einige kolorektale Tumoren teilweise entwickeln, indem sie ein mitochondriales Enzym stilllegen, das normalerweise kleine DNA‑Schnipsel ins Zellinnere gelangen lässt, wo sie wie Leuchtfeuer das Immunsystem alarmieren. Indem ein DNA‑methylierender Partner diese mitochondriale DNA fixiert, halten ACADS‑defiziente Tumoren diese Leuchtfeuer verborgen und entziehen sich so der Immunerkennung. Die Wiederherstellung der ACADS‑Aktivität, etwa durch hypericinähnliche Wirkstoffe, könnte dieses mitochondriale Alarmsystem wieder öffnen, die antitumorale Immunität stärken und die Ansprechrate auf bestehende Immuntherapien verbessern. ACADS, mitochondriales DNMT1 und die STING‑Signalaktivität könnten daher als nützliche Biomarker und therapeutische Ziele im Streben nach wirksameren Behandlungen des kolorektalen Krebses dienen.
Zitation: Yang, F., Wang, M., Hu, S. et al. Short-chain acyl-CoA dehydrogenase initiates mtDNA demethylation and leakage to fuel antitumor immunity in colorectal cancer. Sig Transduct Target Ther 11, 113 (2026). https://doi.org/10.1038/s41392-026-02675-8
Schlüsselwörter: kolorektales Karzinom, Tumorimmunität, mitochondriale DNA, Lipidstoffwechsel, cGAS‑STING‑Signalweg