Clear Sky Science · de
Zielgerichtete Behandlung von FUS (fused in sarcoma): eine neuartige Antisense‑Strategie zur Behandlung der idiopathischen Lungenfibrose
Warum vernarbte Lungen wichtig sind
Die idiopathische Lungenfibrose (IPF) ist eine unerbittliche Lungenerkrankung, bei der feine Lufträume allmählich in starres Narbengewebe verwandelt werden und das Atmen immer schwerer fällt. Die heutigen Medikamente können dieses Vernarbungsverhalten verlangsamen, aber nicht stoppen oder rückgängig machen. Diese Studie untersucht ein neues Ziel namens FUS, ein Protein, das Zellen hilft, mit ihren genetischen Botschaften umzugehen, und prüft, ob das Ausschalten von FUS mit einem speziell entworfenen, DNA‑ähnlichen Strang den Vernarbungsprozess dämpfen und beschädigte Lungen bei der Reparatur unterstützen könnte.
Ein zellulärer Verkehrspolizist gerät außer Kontrolle
FUS ist ein Protein, das normalerweise im Zellkern sitzt und dabei hilft, wie RNA—die Arbeitskopie unserer Gene—verarbeitet und genutzt wird. In Hirnerkrankungen wie ALS kann FUS fehlverhalten, den Kern verlassen, sich im Zellplasma verklumpen und so die normale Zellfunktion stören. Die Autoren fragten, ob ein ähnliches Fehlverhalten die Vernarbung bei IPF antreiben könnte. Sie untersuchten Lungenfibroblasten—die Bindegewebszellen, die Narbenmaterial ablagern—von IPF‑Patienten und von gesunden Spendern. In IPF‑Zellen waren die FUS‑Spiegel insgesamt erhöht und, entscheidend, deutlich mehr FUS im Zytoplasma zu finden als in gesunden Zellen. Mit hochauflösender Elektronenmikroskopie bestätigten sie, dass dieses Protein ungewöhnlich häufig außerhalb des Zellkerns vorkam, was darauf hindeutet, dass seine normale Kontrolle über RNA in fibrotischen Lungen verzerrt sein könnte. 
Wie FUS die narbenbildenden Zellen antreibt
Um herauszufinden, was dieses fehlverhaltende Protein tatsächlich bewirkt, steigerten die Forscher FUS in gesunden Fibroblasten und reduzierten es in IPF‑Fibroblasten. Zusätzlicher FUS veranlasste gesunde Zellen zu schnellerer Teilung, während die Reduktion von FUS in IPF‑Zellen deren Wachstum und Bewegung verlangsamte—zwei Verhaltensweisen, die zentral für die Narbenbildung sind. Das Team nutzte anschließend eine Technik, die Protein–RNA‑Partnerschaften „einfriert“ und die daran gebundenen RNAs ausliest. In IPF‑Fibroblasten war FUS an viele genetische Botschaften gebunden, die Fibrose fördern, einschließlich solcher, die für Kollagen, Wachstumsfaktoren wie TGF‑β und entzündliche Signale kodieren. Mit anderen Worten: FUS fungierte als Knotenpunkt, der ein ganzes Netzwerk pro‑narbiger Botschaften miteinander verband.
Das Signal mit einem Präzisionswirkstoff zum Schweigen bringen
Die Studie testete ein Antisense‑Oligonukleotid namens ION363—einen kurzen, chemisch modifizierten Strang, der so entworfen ist, dass er an FUS‑RNA bindet und deren Abbau auslöst. Wurden IPF‑Fibroblasten mit ION363 behandelt, sanken die FUS‑Spiegel, die Zellen teilten und migrierten langsamer, und zentrale Gene für den Narbenaufbau wurden herunterreguliert. Wichtig ist, dass dieser Effekt nicht auf Zelltod oder erzwungene Seneszenz beruhte; stattdessen schien das Verhalten der Zellen zurückgesetzt zu werden. Als die gleiche Behandlung auf dünne Scheiben von IPF‑Lungengewebe angewendet wurde, die im Labor am Leben erhalten wurden, wurden große Gruppen von Genen, die mit der extrazellulären Matrix, Entzündung und abnormaler epithelialer Auskleidung verknüpft sind, abgeschwächt, während Gene, die mit gesunder Surfactant‑Produktion und alveolärer Funktion assoziiert sind, verstärkt wurden. Die Behandlung verringerte zudem die Kollagenfärbung und erhöhte Marker funktioneller oberflächenbildender Lungenzellen, was auf eine Verschiebung von Narbenbildung hin zu Reparatur hindeutet.
Schadhaftes Lungenbläschenwachstum unterstützen
Da die kleinen, die Lungenbläschen auskleidenden Zellen, bekannt als Typ‑II‑Alveolarzellen, für die Lungenreparatur entscheidend sind, bauten die Forscher dreidimensionale „Alveolosphären“ aus Patienten‑Zellen, um miniaturisierte Lungeinheiten zu modellieren. In Kulturen von IPF‑Patienten überleben diese Strukturen normalerweise schlecht. Mit ION363‑Behandlung entstanden mehr Alveolosphären, sie wuchsen größer und zeigten erhöhte lysosomale Aktivität—ein Kennzeichen aktiver Erneuerung. Detaillierte Färbungen zeigten mehr Zellen mit Markern reifer gasaustauschender Zellen, was darauf hindeutet, dass das Stilllegen von FUS nicht nur Fibroblasten beruhigte, sondern auch das verletzte Epithel dazu anregte, eine gesündere Oberfläche wieder aufzubauen. 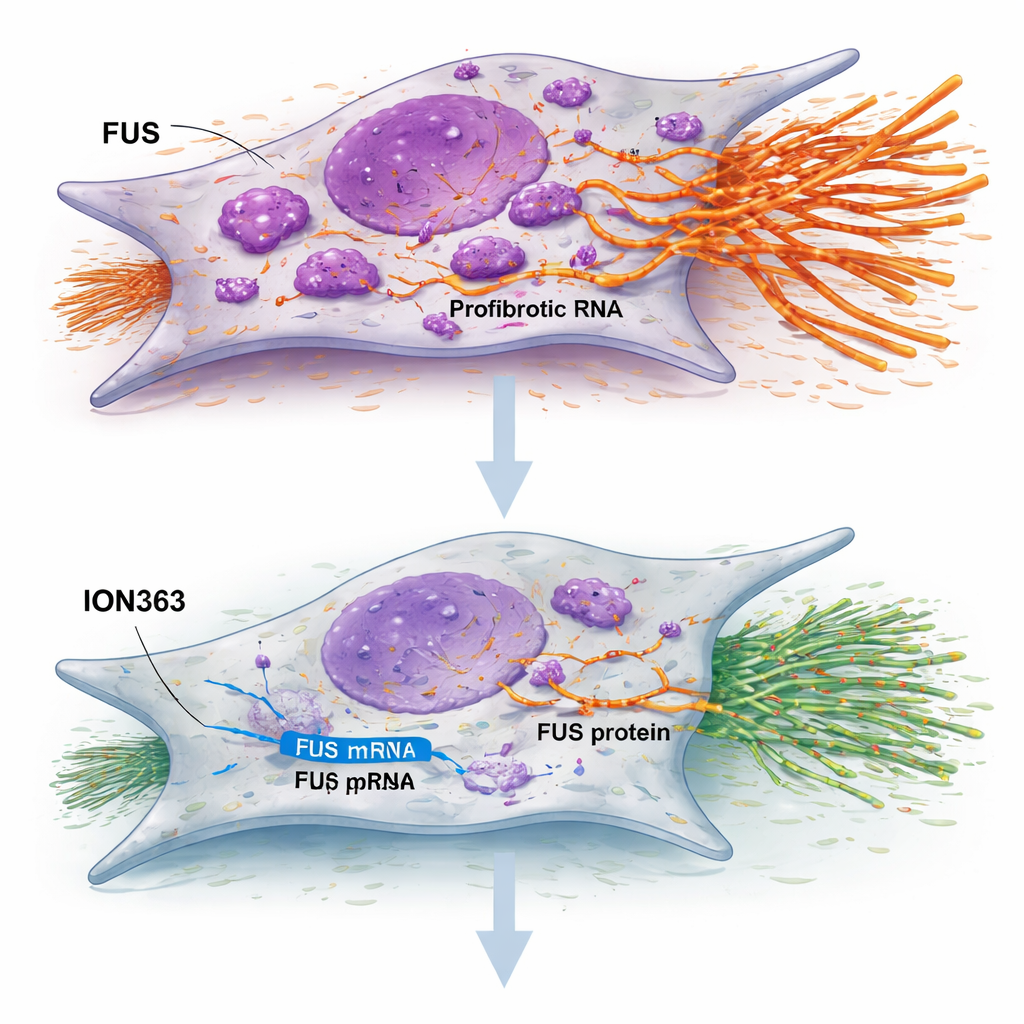
Was das für Patienten bedeuten könnte
Zusammengefasst zeichnet die Arbeit FUS als Master‑Schalter in der IPF, der überaktive narbenbildende Fibroblasten mit einer versagenden Reparatur der empfindlichen Lufträume verbindet. Durch das Herunterregeln von FUS mit einem zielgerichteten Antisense‑Wirkstoff konnten die Forscher profibrotische Genprogramme reduzieren, Kollagenansammlungen mildern und Regeneration in patientenabgeleiteten Lungenmodellen fördern. Obwohl dieser Ansatz noch im Laborstadium ist und sorgfältige Tests in Tiermodellen und klinischen Studien erfordert, legt er nahe, dass IPF eines Tages nicht nur durch Verlangsamung der Vernarbung behandelt werden könnte, sondern durch ein direktes Wieder‑Ausbalancieren der zellulären Programme, die Lungenverletzung und Reparatur steuern.
Zitation: Katariya, B.B., Chillappagari, S., Arnold, L. et al. Targeting fused in sarcoma (FUS): a novel antisense strategy for treating idiopathic pulmonary fibrosis. Sig Transduct Target Ther 11, 70 (2026). https://doi.org/10.1038/s41392-026-02585-9
Schlüsselwörter: idiopathische Lungenfibrose, Antisense‑Oligonukleotid, FUS‑Protein, Lungenfibrose, Alveoläre Reparatur