Clear Sky Science · ar
نموذج فأر قابل للحياة مع RIPK3 عديم الكيناز D143N يكشف دوره الحامل في تحفيز اضطراب التهابي مستحث بواسطة TNF
لماذا تهم هذه الدراسة على الفئران في موضوع الالتهاب
العديد من الأمراض الشديدة، من العدوى المميتة إلى نوبات الأمراض المناعية الذاتية، لا يقودها فقط الجراثيم أو الجينات بل أيضاً الالتهاب المفرط الذاتي للجسم. يُنظر إلى بروتين يسمى RIPK3 منذ وقت طويل كمنفِّذ رئيسي لشكل عنيف من موت الخلايا يغذي هذا الالتهاب، مما يجعله هدفًا دوائيًا جذابًا. لكن لـ RIPK3 أدوار أخرى داخل الخلايا أقل فهماً. تصف هذه الدراسة نوعًا جديدًا من الفئران المخبرية يفصل بوضوح بين نشاط RIPK3 القاتل ودوره الإشاري «الحامل»، كاشفة كيف تساهم كل وظيفة في الالتهاب ومشيرة إلى استراتيجيات علاجية جديدة.
طريقتان يمكن أن يعمل بهما بروتين الموت
يمكن للخلايا أن تموت بطرق منظمة أو فوضوية. في موت الخلايا المرتب أو «الصامت»، يعيد الجسم تدوير أجزاء الخلية بهدوء دون إنذار كبير. في شكل أكثر فوضوية يسمى النيكروبتوز، تنفجر الخلايا وتنسكب محتوياتها، مما يثير استجابات مناعية قوية. RIPK3 مركزي في النيكروبتوز: عند تنشيطه، يفعل بروتينًا آخر يُحدث ثقوبًا في غشاء الخلية. مع ذلك، أشارت أعمال سابقة إلى أن RIPK3 يمكن أن يساعد أيضًا في تحفيز الانتحار الخلوي المعتمد على الكاسباس ويعزز الإشارات الالتهابية حتى دون قتل الخلايا. كان من الصعب تفكيك هذه الأدوار لأن أشكال RIPK3 غير النشطة الموجودة سابقًا إما كانت تقتل الأجنة أو تقلل بشكل كبير من مستويات البروتين، مما صعّب دراسة سلوكه الحامل الطبيعي.
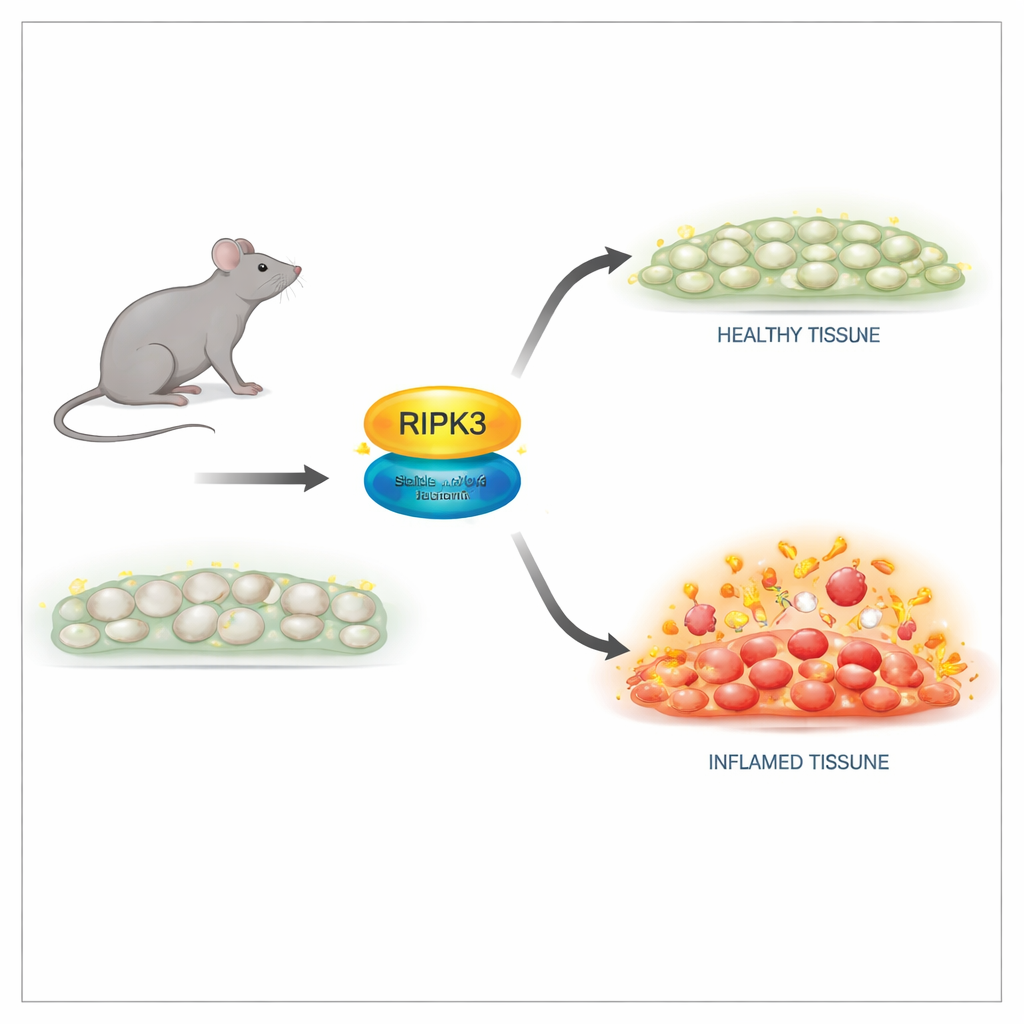
طريقة أكثر أمانًا لإيقاف وظيفة القتل
قام الباحثون بتعديل جيني للفئران ليحمل تغييرًا دقيقًا في بروتين RIPK3 عند موقع واحد، أطلقوا عليه D143N، يوقف نشاطه الإنزيمي بينما يحافظ على بنيته. في خلايا هذه الفئران، بدت مستويات بروتين RIPK3 وبنية الأنسجة طبيعية، وُلدت الحيوانات ونمت تمامًا مثل إخوتها الأصحاء. الأهم من ذلك، كانت خلايا النسخة D143N مقاومة تمامًا لمجموعة من مثيرات النيكروبتوز، بما في ذلك إشارات عامل نخر الورم (TNF) ومستقبلات شبيهة بالتول ولعدوى فيروسية. لم يعد RIPK3 المتحول قادرًا على تفعيل الشريك السفلي أو تشكيل المركب المدمر اللازم لانهيار الغشاء، ومع ذلك لم يسبب هذا التحور انتحارًا مبرمجًا عفويًا، متجنبًا التأثيرات الجانبية القاتلة التي وُجدت في طفرات RIPK3 الأقدم.
فصل التطور عن المرض
أحد أدوار RIPK3 المعروفة جيدًا يظهر في الأجنة التي تفتقر إلى بروتين رئيسي آخر، الكاسباس-8: بدون الكاسباس-8، يقتل النيكروبتوز المدفوع بـRIPK3 الجنين. في هذه الدراسة، أنقذ إدخال نسخة D143N من RIPK3 هذه الفئران التي كانت غير قابلة للحياة تمامًا. تطورت عاديًا وكانت خصبة، مما يثبت أن نشاط القتل لـRIPK3 غير ضروري للتطور الطبيعي طالما أن بنيته محفوظة. ومع ذلك، عند تحدي الفئران البالغة بجرعات عالية من TNF لإحداث متلازمة التهابية شبيهة بالصدمة، تغيرت الصورة. الحيوانات التي تفتقر تمامًا إلى RIPK3 كانت محمية بقوة من الموت وتلف الأنسجة وجزيئات الالتهاب في الدم. أما الفئران الحاملة لنسخة D143N، بالرغم من افتقارها للنيكروبتوز، فكانت محمية جزئيًا فقط. هذا أشار إلى أن دور RIPK3 غير القاتل، كقالب حامِل، لا يزال يساهم في دفع الالتهاب.
إشارات الحامل التي تأجج الالتهاب
لفهم هذه المساهمة غير القاتلة، فحص الفريق نشاط الجينات في أمعاء الفئران المعالجة بـTNF. في الحيوانات الخالية من RIPK3، كانت العديد من الجينات الالتهابية مُخفّضة بشدة. أما في فئران D143N، فكان الكبت أضعف، وبقيت الجينات المرتبطة بالإنترفيرون والاستجابات المناعية الفطرية أكثر نشاطًا. على مستوى البروتين، فعّل TNF بقوة مسارات الإشارة JAK–STAT1 وERK في الفئران الطبيعية وD143N، لكن هذا التفعيل كان شبه غائب عندما حُذف RIPK3 تمامًا. أظهر ذلك أنه حتى من دون وظيفة القتل، تساعد الوجود المادي لـRIPK3 في مركبات الإشارة على نقل إشارات TNF إلى برنامج مُحرّض للالتهاب عبر JAK–STAT1.
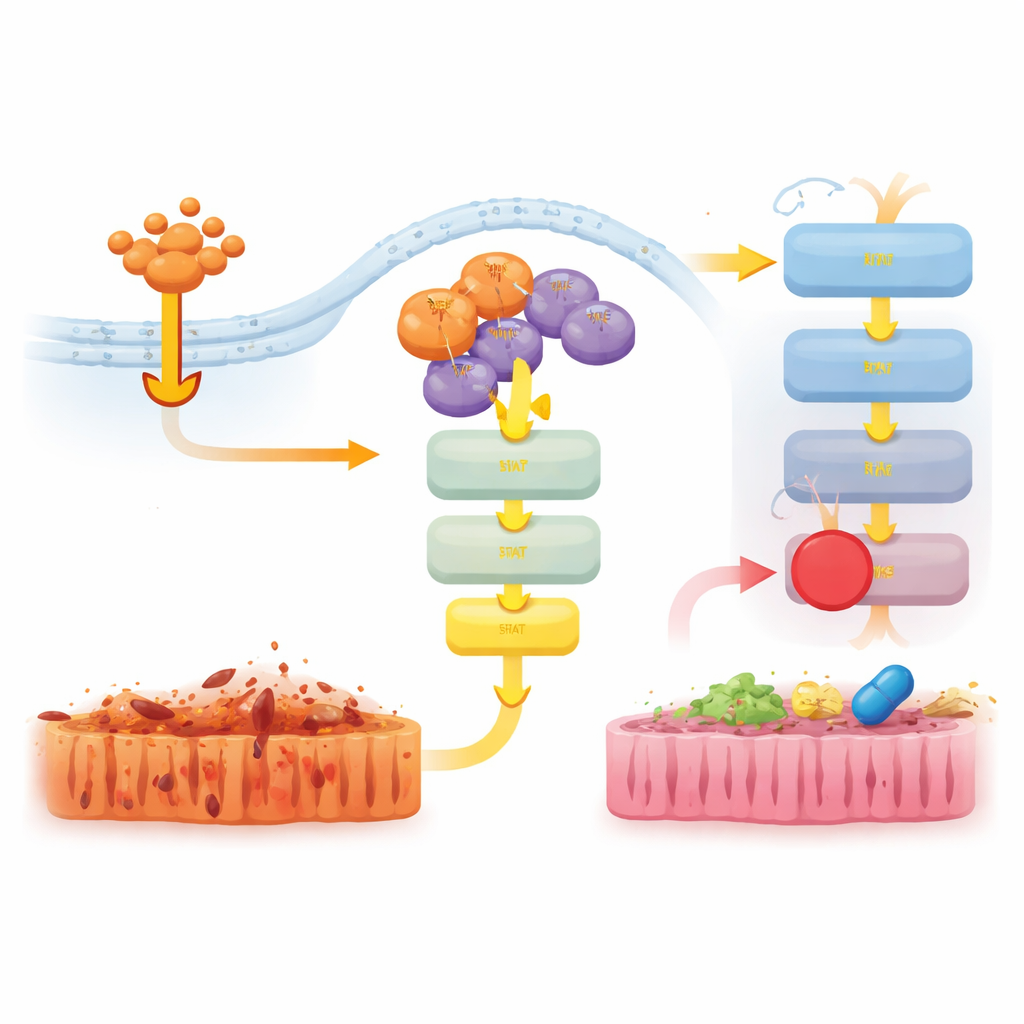
خفض الإشارات الضارة بأدوية موجهة
ثم اختبر الباحثون ما إذا كان حجب هذه المسارات السفلى يمكن أن يخفف المرض في فئران D143N أثناء صدمة مستحثة بـTNF. أدى علاج الحيوانات بمثبط JAK1/2، وليس مثبط ERK، إلى تقليل فقدان درجة الحرارة وخفض مستويات الجزيء الالتهابي IL-6 وتخفيف تلف نسيج الأمعاء وموت الخلايا. كما حمى مثبط آخر يستهدف بروتينًا مختلفًا، RIPK1، الفئران بقوة وقمَع تنشيط JAK–STAT1 وERK. تقترح هذه النتائج مجتمعة أن وظيفة الحامل لـRIPK3 تتعاون مع RIPK1 لتنشيط JAK–STAT1 ودفع الالتهاب، وأن مقاطعة هذه الإشارة يمكن أن تقلّل إصابة الأنسجة حتى عندما يكون النيكروبتوز محظورًا بالفعل.
ما الذي يعنيه هذا للعلاجات المستقبلية
لسنوات اعتُبر RIPK3 بشكل أساسي مفتاحًا لشكل سام من موت الخلايا، وركّز تطوير الأدوية على إيقاف نشاطه الإنزيمي. تُظهر هذه الدراسة أن ذلك قد لا يكون كافياً: يمكن أن يظل RIPK3 يعمل كمنصة فيزيائية تضخّم الإشارات الالتهابية عبر JAK–STAT1، مساهماً في الصدمة وتلف الأنسجة. يكشف نموذج الفأر الجديد D143N عن هذه الأدوار المزدوجة بوضوح غير عادي، مما يوفر أداة قوية لدراسة متى وكيف تهم كل وظيفة في أمراض مختلفة. وللمرضى، يشير العمل إلى أن الجمع بين أدوية تستهدف RIPK3 أو RIPK1 مع مُثبِّطات JAK–STAT1 قد يهدئ الالتهاب الضار بشكل أكثر فعالية في الحالات التي يقودها TNF والسيتوكينات المرتبطة به.
الاستشهاد: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
الكلمات المفتاحية: RIPK3, النيكروبتوز, الالتهاب, صدمة TNF, JAK-STAT1