Clear Sky Science · zh
使用化学储层的能效科学计算
为何将化学转化为计算很重要
现代超级计算机在模拟气候、设计新药或训练人工智能时消耗大量电力。随着我们接近传统芯片的物理极限,在每瓦特性能上继续提升变得越来越困难且昂贵。本文探讨了一条根本不同的途径:把真实的化学反应作为科学计算的驱动引擎。通过将分子及其相互作用视为计算机的运动部件,作者勾勒出未来机器如何以远低于当今数字硬件的能耗来求解复杂方程。
从活细胞到化学计算器
活细胞是卓越的问题解决者。它们不断协调成千上万的反应以适应、成长和生存,同时消耗的能量却极其有限。这一行为的核心是化学反应网络——相互连接的反应,其速率和浓度随时间变化。这些网络可以用常微分方程来描述,这与用来建模从流行病到湍流的一切现象所采用的数学语言相同。本研究的洞见在于:既然化学已经遵循这些方程,我们或可直接利用化学来执行科学家当前在硅芯片上进行的计算。
方程如何变成反应网络
作者介绍了 ChemComp,这是一个将微分方程系统系统性转换为抽象反应网络的软件框架。ChemComp 使用现代编译器技术,将数学问题分解为可由理想化反应表示的模式,然后将它们组织成具有明确定义的物种、连接和速率的网络。这些抽象反应尚不对应真实分子,但它们构成了化学计算机的蓝图。该框架还可以在生化反应数据库中搜索,寻找行为相似的真实反应模板,并偏好在实验环境中更实用、安全且可能更节能的选项。
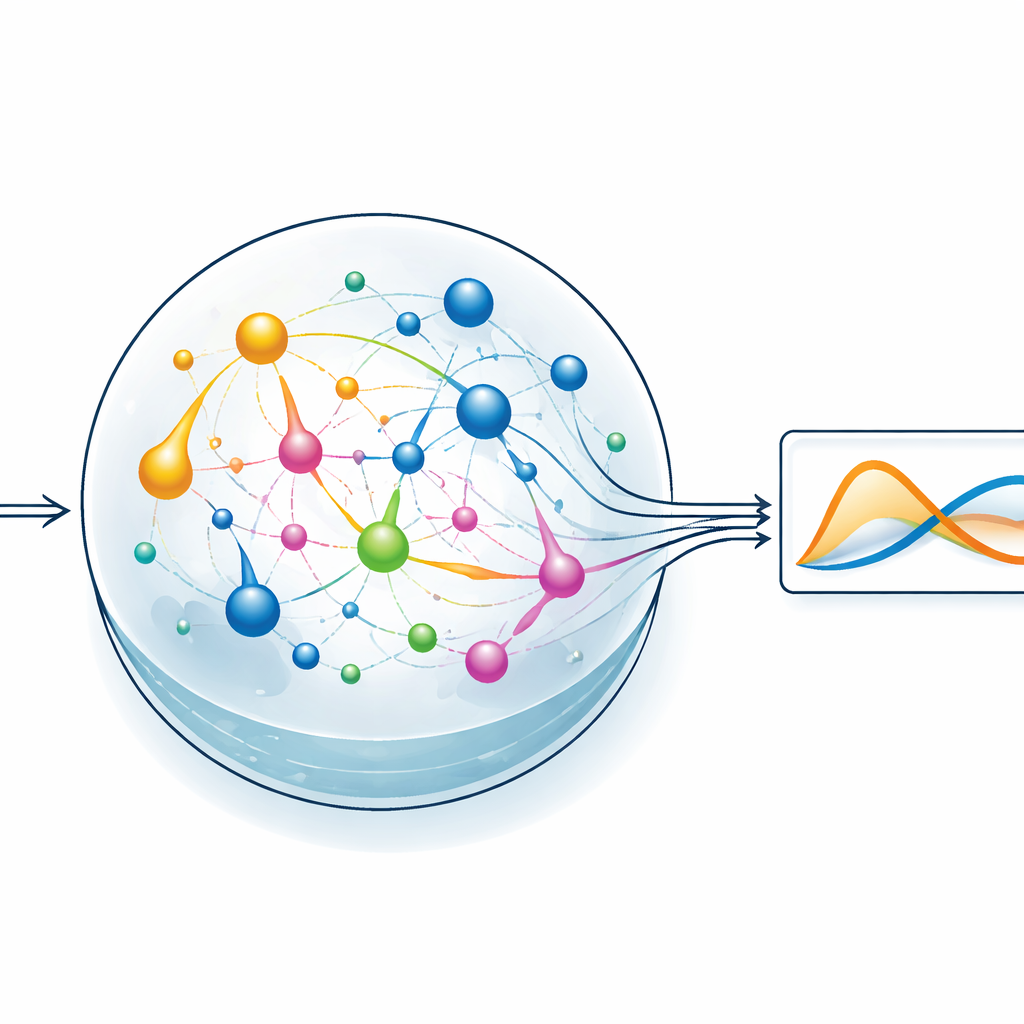
让化学储层承担繁重工作
为检验该想法,团队聚焦于一种称为储层计算的机器学习风格。在这种方法中,一个固定的、动态的系统将输入信号转换为丰富而纠结的内部活动模式,只有一个简单的读出层需要经过训练以产生期望输出。在 ChemComp 的实现中,储层是一个在良好搅拌容器内的反应集合;化学物质浓度的变化构成内部状态。作者将一种经典的双变量系统——Sel’kov–Schnakenberg 模型(最初用于研究代谢振荡)——编译成候选反应网络。然后他们模拟这些网络在通过进出容器的化学流驱动下随时间的响应,并用基本的线性回归把浓度轨迹组合成对目标解的近似。
测试简单与更复杂的化学网络
研究者比较了两种候选储层:一种只有两个化学物种和两条反应,另一种有五个物种和五条反应。两者都给定合适的初始浓度和流速,然后模拟运行。即便是较小的系统也能大致再现目标方程的振荡行为,但较大的网络表现明显更好,在训练和测试期间均降低了误差。通过扫描不同的初始浓度和反应速率常数,作者绘制出化学系统最接近所需动力学的区域。每一条反应在曲线拟合问题中实际上都像一个基函数:可用反应越多,越容易逼近复杂行为,但代价是系统复杂度增加。
走向实验室中的低能耗计算
除了模拟外,文章还展望了实用装置。讨论了反应选择如何在能耗、通过酶或催化剂的可控性以及实时测量关键物种(例如通过光学或电化学方法)的能力之间取得平衡。作者建议未来的微流控平台可以承载精心选择的反应网络,具备空间上的输入控制和内置感测。尽管从将方程映射到真实化学到处理噪声与测量极限,仍有许多工程挑战,但研究表明,适度的反应系统已经能够模拟耦合微分方程的解。对非专业读者而言,核心信息是:化学本身可以作为模拟计算机,从而开辟一条利用自然数十亿年完善的能效过程来进行科学计算的道路。
引用: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
关键词: 化学计算, 节能计算, 储层计算, 化学反应网络, 常微分方程