Clear Sky Science · zh
用于异相催化反应性的端到端框架
为何更快的催化剂设计至关重要
现代社会依赖催化剂来制造燃料、塑料、化肥以及无数日常用品。然而,要找到更好的催化剂常常像大海捞针,因为每种材料可以同时促进数千种可能的微观反应。本文介绍了 CARE——一种新的计算框架,它结合智能规则与机器学习,能够比以往更快、更全面地绘制并模拟这些错综复杂的反应网络。通过如此做法,它有望引导更清洁的能源技术和更高效的化学过程,同时大幅降低计算成本。
解开拥挤的反应路径
在固体催化剂表面,进入的分子并非沿着单一整齐的路线从反应物变为产物。相反,它们穿过由短寿命中间体和相互竞争路径组成的迷宫。传统计算方法依赖人为直觉来挑选有限的一组可能步骤,然后用量子计算评估其能量。这在小型网络上可行,但随着体系复杂度增长很快就失效,会忽视那些罕见但可能主导长期活性、失活或选择性的通路。CARE 通过从简单构建规则自动构建非常大的反应网络来应对这一挑战,确保包括碳、氢和氧之间所有合理的断键与成键事件,即便是那些化学家通常可能会舍弃的路径。
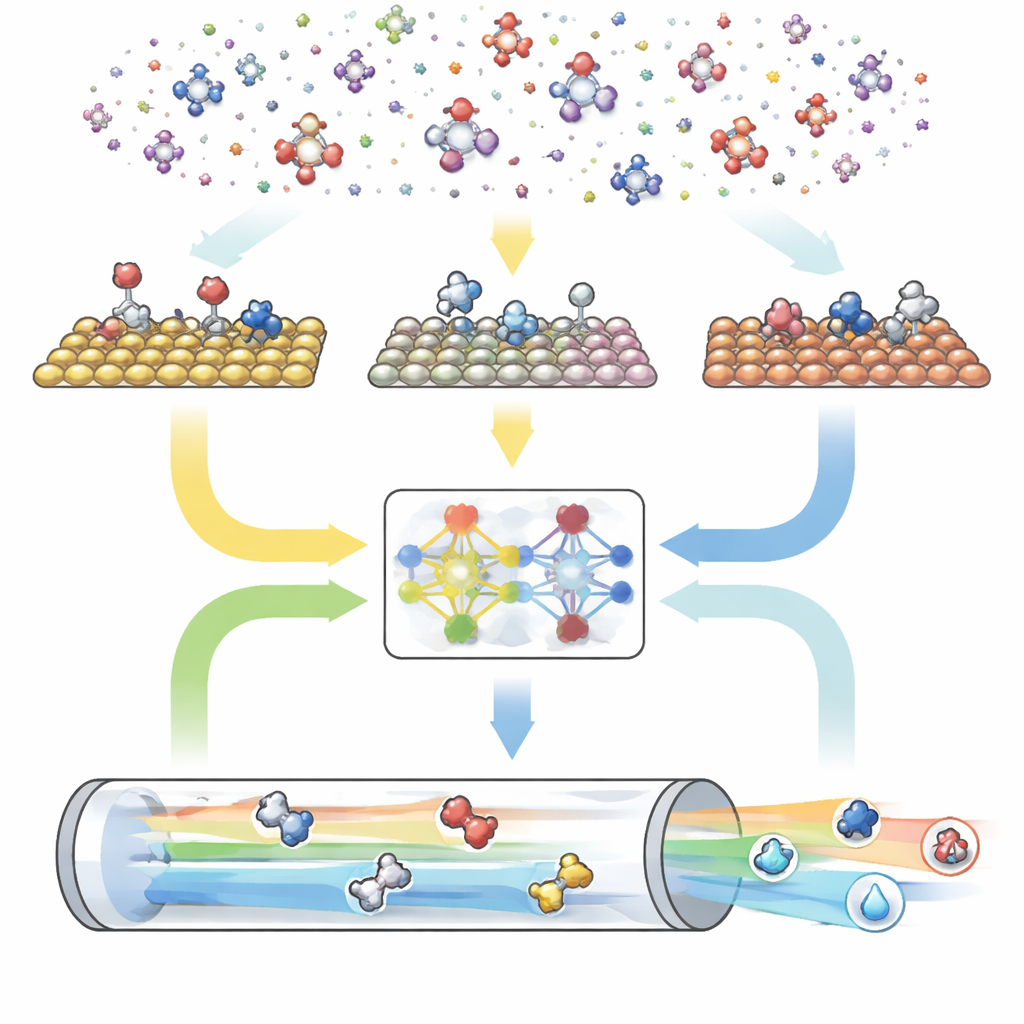
用于反应的三部分数字引擎
CARE 作为一个端到端流水线构建,由三个主要模块组成。首先,基于规则的生成器通过选择最大碳原子数和氧原子数,然后应用简单模板来创建所有匹配的分子及其吸附态,定义了“化学空间”。其次,能量评估模块调用现代机器学习模型——特别是名为 GAME-Net-UQ 的图神经网络——来估算许多金属表面上中间体和过渡态的能量。该模型将每个结构视为原子与键的网络,返回能量与不确定性,精度在几十分之一电子伏范围内,同时保持轻量和快速。第三,微观动力学求解器利用这些能量计算在现实温度、压力、电压与 pH 条件下所有反应如何共同进行,预测总体反应速率、表面覆盖度与产物选择性。
实际测试:燃料分子与气候化学
为证明 CARE 不只是理论练习,作者将其应用于三个工业相关且难度逐步增加的问题。对于甲醇分解——这对氢贮存很重要——他们生成了一个适度的网络并在多种金属催化剂和晶面上评估它。CARE 再现了熟悉的活性“火山”趋势,并正确地将钌识别为表现最好的催化剂之一,这与实验一致,但所需计算时间只是完整量子计算的极小一部分。接着,他们研究了铜上二氧化碳的电化学转换,聚焦三碳产物如 1-丙醇和丙烯如何生成。通过包含考虑质子、电子和溶液条件的特殊步骤,CARE 捕捉到 pH 与外加电压如何改变路径,并正确预测 1-丙醇比丙烯更有利,这与先前的详细研究相呼应。
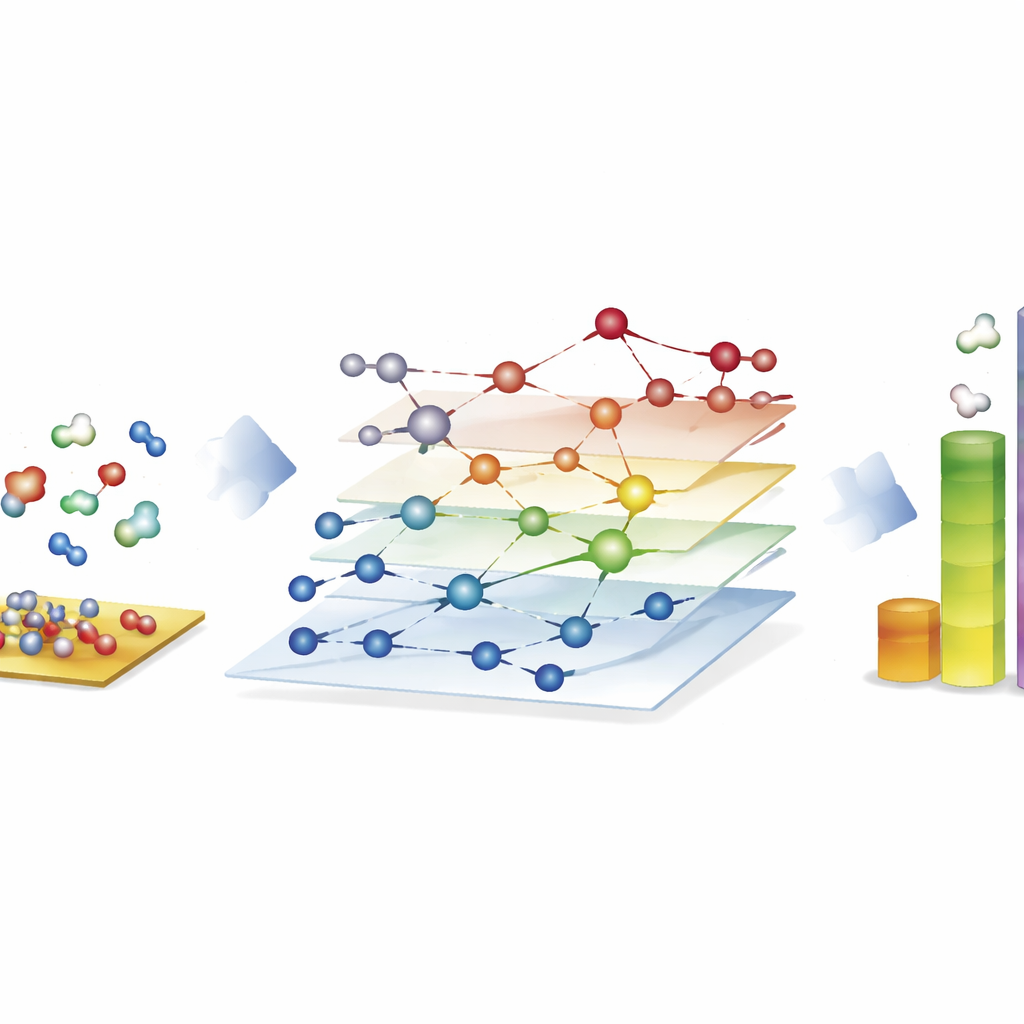
为合成燃料探索巨大的反应网络
最引人注目的示例来自费-托(Fischer–Tropsch)过程,该过程将一氧化碳和氢的混合物转化为用于燃料和化学品的长链烃。在这里,作者构建了近 4 万种表面物种和约 37 万个基本反应的网络——远远超出传统基于量子研究的全面探索能力。利用 CARE,他们在标准硬件上仅用几小时就评估了钴、铁、镍和钌表面上所有中间体和关键反应势垒,与直接量子计算相比速度提高了约百万倍。基于这些网络的微观动力学模拟再现了已知趋势:钴和铁更倾向形成更长的烃链,铁通过副反应产生更多二氧化碳,而镍则倾向于更强的氢化。尽管一些细节(例如甲烷产率)仍不完美,该框架揭示了主导链增长的成键步骤,并指出模型仍需改进的关键环节。
这对未来催化剂意味着什么
对非专业读者而言,核心信息是 CARE 提供了一种实用途径来探索此前难以企及的催化表面上的巨大反应空间。通过自动化网络生成、引入用于量子能量的快速机器学习“替代”模型并高效求解所得动力学,它可以对候选催化剂进行排序、识别有前途的操作条件并发现出乎意料的路径,同时显著减少人为偏见和计算开销。作者也指出了尚存挑战——例如更好地处理拥挤表面、溶剂效应以及更大的网络——但这项工作指向了一个未来:计算机能快速筛查复杂反应(从二氧化碳还原到塑料回收和生物质升值),引导实验朝最有希望的想法发展,而不是仅凭反复试验来发现。
引用: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
关键词: 异相催化, 反应网络, 机器学习, 微观动力学建模, 费-托合成