Clear Sky Science · zh
用于生成多样蛋白构象集的基于局部感知模态对齐的条件扩散
为什么蛋白质运动很重要
细胞中的蛋白质并非静止的雕塑;它们更像持续改变形状的微小柔性机器。这些形状变化可以控制酶如何催化反应、受体如何响应药物以及信号如何在细胞中传递。然而,我们熟悉的大多数蛋白质图像只显示单一的“快照”结构,遗漏了实际上存在的丰富构象集合。本文介绍了 Mac-Diff,一种人工智能方法,能够快速为给定蛋白生成大量逼真的构象,帮助科学家不仅看见蛋白的样子,还能察觉其如何“呼吸”和运动。
从单张快照到动态构象集
几十年来,研究人员依赖耗时的实验或长时间运行的分子动力学模拟来探索蛋白质运动,这些方法往往又慢又昂贵。像 AlphaFold2 这样的突破性工具现在可以直接从氨基酸序列预测蛋白的最可能三维结构,但通常只返回一种或少数几种优先构象。许多蛋白质,尤其是参与信号转导和变构调控的蛋白,天然占据多个松散定义的状态。作者认为,要理解这类蛋白如何真正工作——并设计能结合不那么明显、短暂构象的药物——我们需要一种能够生成整套合理构象的方法,而不仅是一种最佳猜测。
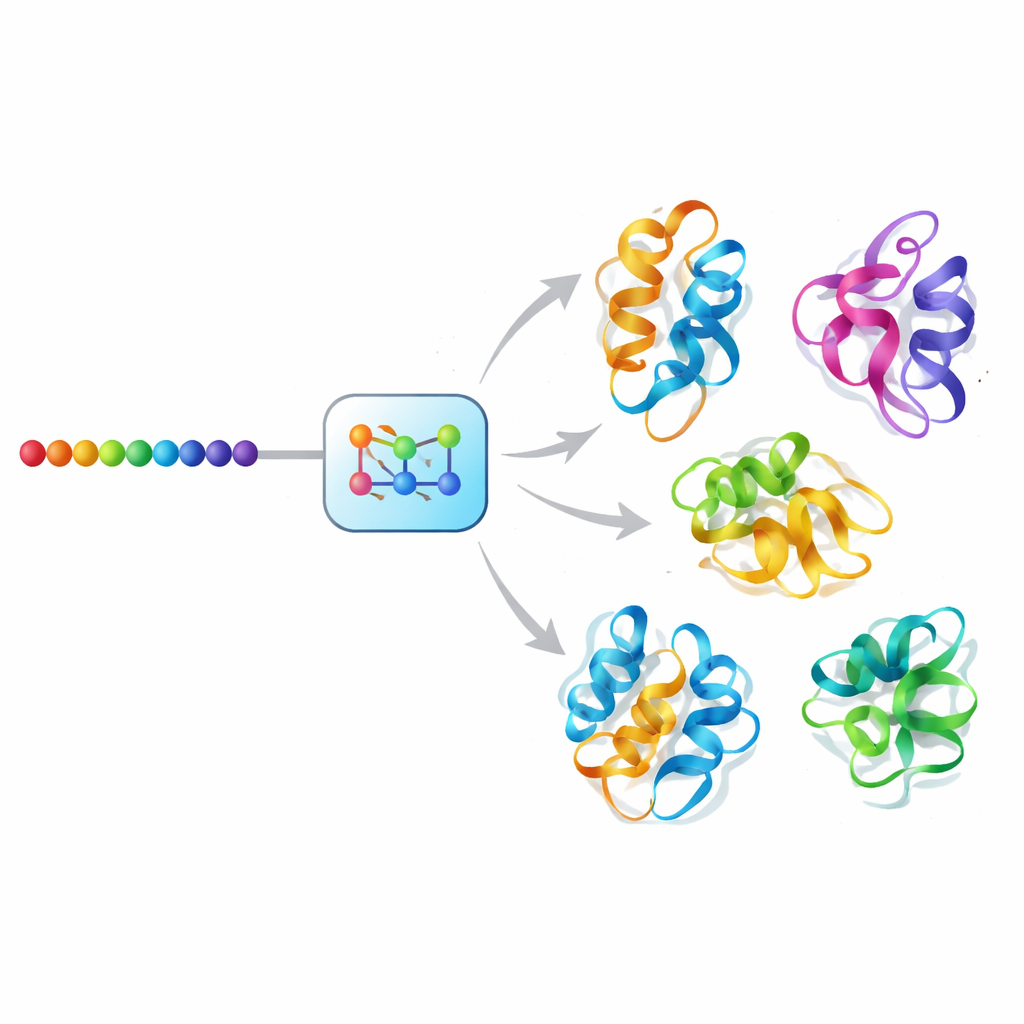
一种用于蛋白质运动的人工智能“扩散”方法
Mac-Diff 使用一种扩散式生成模型来应对这一挑战,这类人工智能方法推动了图像合成方面的近期飞跃。Mac-Diff 并非对照片去噪,而是对蛋白质骨架的抽象几何描述去噪。该模型将蛋白表示为残基间成对关系的网格——这些关系包括不受整体旋转或平移影响的距离和角度。在正向过程中,系统逐步向这些几何模式添加噪声,直至它们类似随机静态。在反向过程中,模型学习逐步去除噪声,以蛋白的氨基酸序列为引导,直到出现连贯且与三维兼容的几何结构,然后可由标准构建软件转换为完整的原子模型。
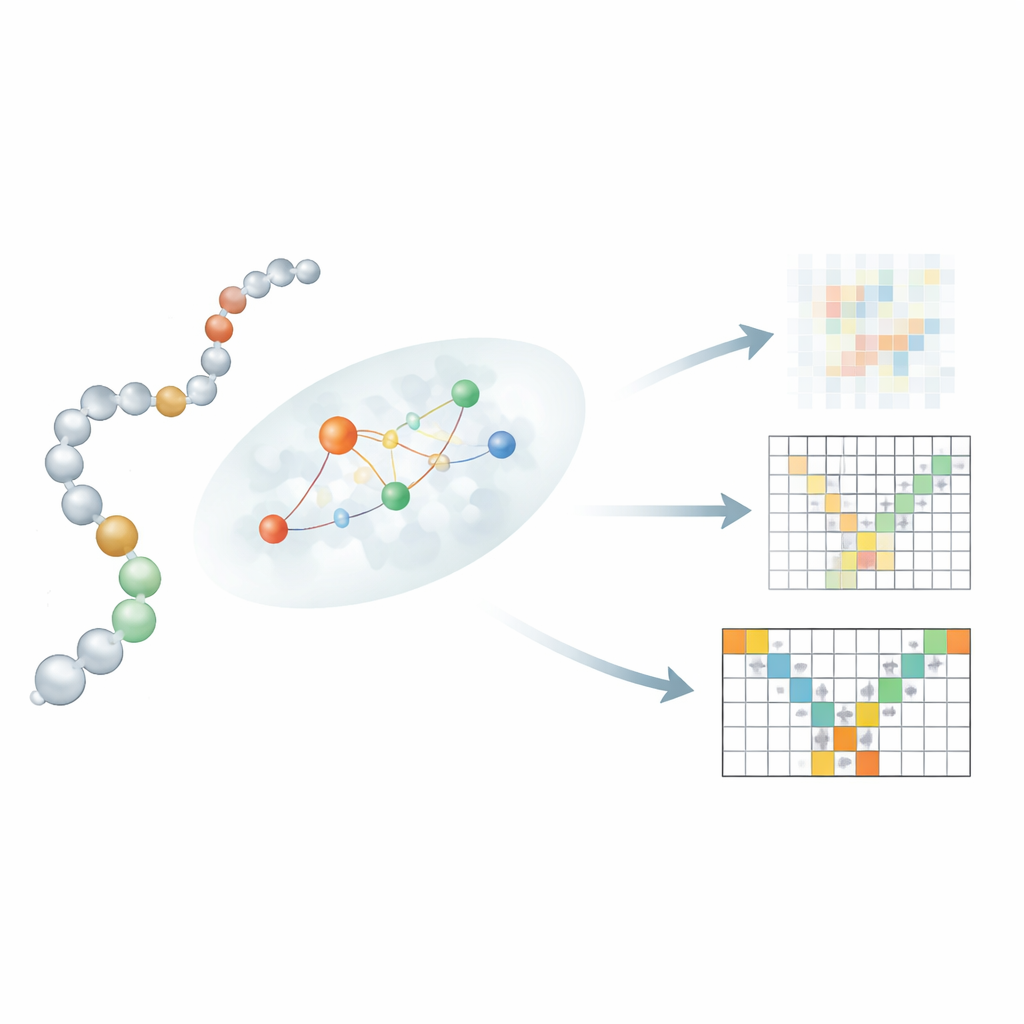
让序列在局部与结构对话
关键创新在于 Mac-Diff 如何将线性残基序列与其三维邻居连接起来。像文本到图像模型那样让每个残基关注所有其他残基,会模糊重要的物理约束。相反,作者引入了一种“局部感知”注意力机制,使每个残基专注于一个小而可能的相互作用邻域。为估计这些邻域,Mac-Diff 使用三种要素:一种名为 ESM-2 的蛋白语言模型,用于编码每个残基的生化上下文;提示哪些残基对可能相近的接触图;以及一个偏好链上接近残基的简单规则。这些信号被组合起来,以便在去噪过程中模型优先利用那些在物理上更可能成为相互作用伙伴的残基,从而增强其重建现实、柔性的结构的能力。
与长时模拟和构象可变蛋白的对比测试
研究者在两个严苛方向上测试了 Mac-Diff。首先,他们考察它是否能重现长时间、精确计算的分子动力学模拟中观察到的广泛构象分布,这些模拟包括快折叠蛋白以及一个经典基准蛋白 BPTI。在若干比较生成构象集与模拟数据统计特性的指标上——例如蛋白内距离分布和整体紧凑度——Mac-Diff 与其它竞争的人工智能方法相比达到或超越了它们,同时生成了更广泛的构象多样性。它捕获了模拟中识别出的多数关键“亚稳”状态,并以高相关性重现了残基级的柔性模式,表明其构象集在全局折叠和局部摆动两方面都具有现实性。
揭示隐藏的功能状态
其次,团队用已知在执行功能时会采取非常不同形态的蛋白对 Mac-Diff 提供挑战,包括在能量代谢中在开/闭态之间切换的腺苷酸激酶,以及一组经过挑选、每个都有两个实验证定构象的40个蛋白。Mac-Diff 每个蛋白仅生成100个候选结构——远少于典型模拟轨迹——但仍以良好的几何一致性找回了大多数已知状态。例如在腺苷酸激酶中,它生成了与晶体结构高度相似的开与闭构象,而若干流行方法往往偏向于仅生成一种状态。该模型在可比硬件上运行速度也比传统模拟快约一千倍,使系统性地探索构象多样性变得更加可行。
这对生物学和医学意味着什么
通俗来说,Mac-Diff 把蛋白的序列变成了一个合理姿态的画廊,而不是一幅单一肖像,并且它考虑了哪些部分在三维中更可能相互推动或抓握。通过准确且高效地抽样这些构象集,该方法提供了一种探查微妙构象变化如何影响功能、发现罕见但重要构象以及搜索仅在短暂状态中出现的药物结合口袋的途径。尽管它尚未捕捉到模拟所提供的完整时间序列电影,Mac-Diff 将蛋白的动态景观带入了更多系统的可及范围,有望为结构生物学、药物设计和蛋白质工程带来新见解。
引用: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
关键词: 蛋白质动力学, 扩散模型, 构象集, 变构蛋白, 药物发现