Clear Sky Science · zh
从纳米颗粒到单原子位点:机器学习引导的能源相关催化剂设计
更聪明的配方,清洁能源更近一步
设计更好的催化剂——那些加速化学反应的微小材料——对于更清洁的燃料、更便宜的电池和更环保的工业至关重要。但找到合适的“配方”长期以来一直是一个缓慢的试错过程。本文阐述了推动现代人工智能的技术——机器学习——如何正在改变这一探索。通过教会计算机在海量数据中识别模式,科学家现在能够更快地锁定有前途的催化剂设计,尤其是针对由纳米颗粒乃至单个金属原子构成的前沿材料。
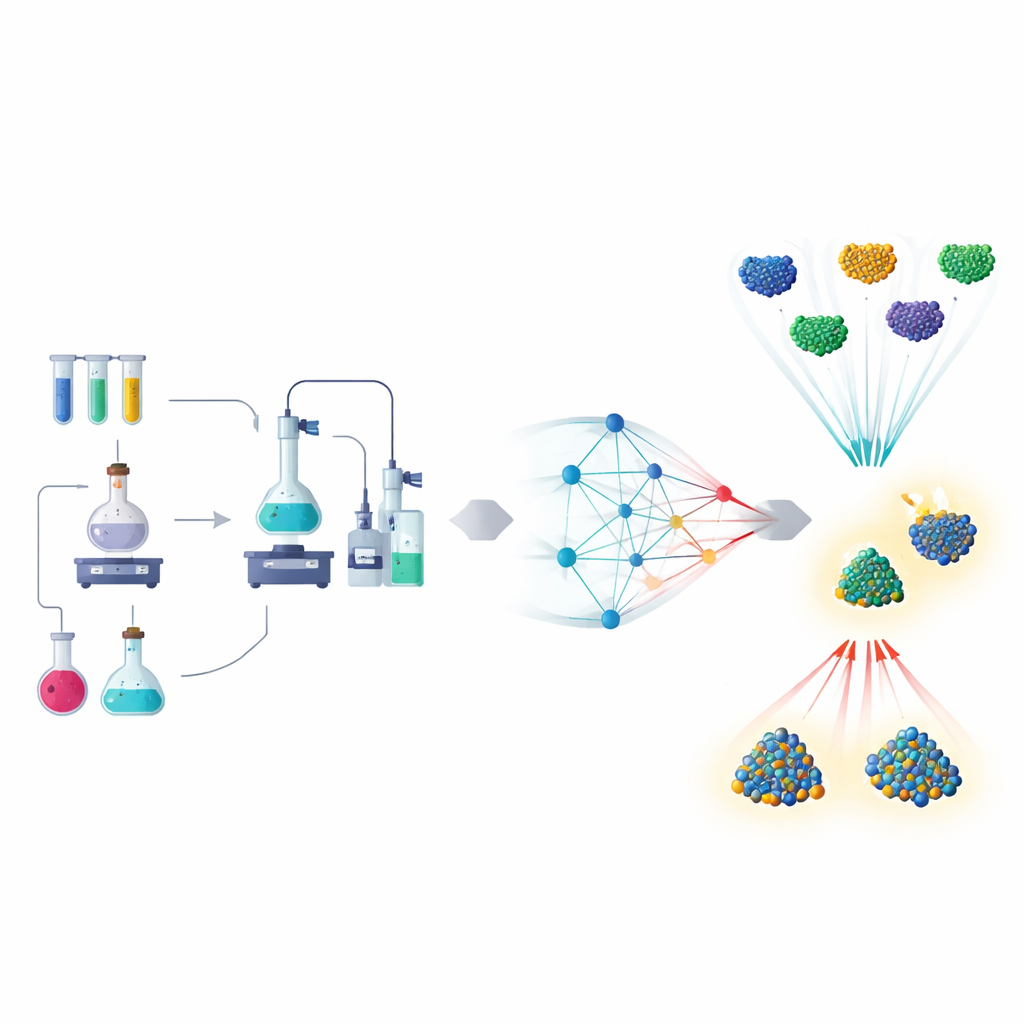
从试错到数据驱动的发现
传统的催化剂研究类似于没有明确食谱的烹饪:换一种金属、换一种载体、调整温度,然后测试再重复。论文描述了这种方法如何被机器学习模型重塑,这些模型从实验和量子级模拟中学习。模型可以在不做每一项实验的情况下预测催化剂的行为——例如关键分子的吸附强度、反应速率或材料的寿命。因此,科学家可以在计算机上筛选数千种可能性,并将宝贵的实验时间留给最有希望的候选者。
纳米颗粒:理想的试验场
早期的大部分进展来自纳米颗粒催化剂:金属原子组成的微小簇可以执行分解水或转化二氧化碳等反应。在这里,机器学习使用诸如颗粒尺寸、表面结构和成分等简单输入来预测性能。通过消化多年实验和模拟积累的数据,这些模型可以建议下一步尝试的合金组合或需要探索的反应条件。由这些预测驱动的自动化机器人现在能在很少人工干预的情况下运行数百次实验,从而极大地加速了能源和环境技术更优材料的发现。
单原子为何如此特殊
综述进一步聚焦于单原子催化剂,即将单个金属原子锚定在固体载体上的体系。这类催化剂有个诱人承诺:每个金属原子都可以是活性的,从而把昂贵元素(如铂或铱)的用量降到最低。但因为每个原子所处的局部环境各不相同,其行为对与邻近原子的键合方式极为敏感。作者展示了机器学习如何帮助解码这种复杂性。通过向模型输入简单的数值描述符——例如金属的电子数、其吸电子能力或与邻居的配位方式——研究者可以绘制出结构如何控制关键反应(如析氧、燃料电池过程、氮固定和二氧化碳还原)的活性、选择性与稳定性。
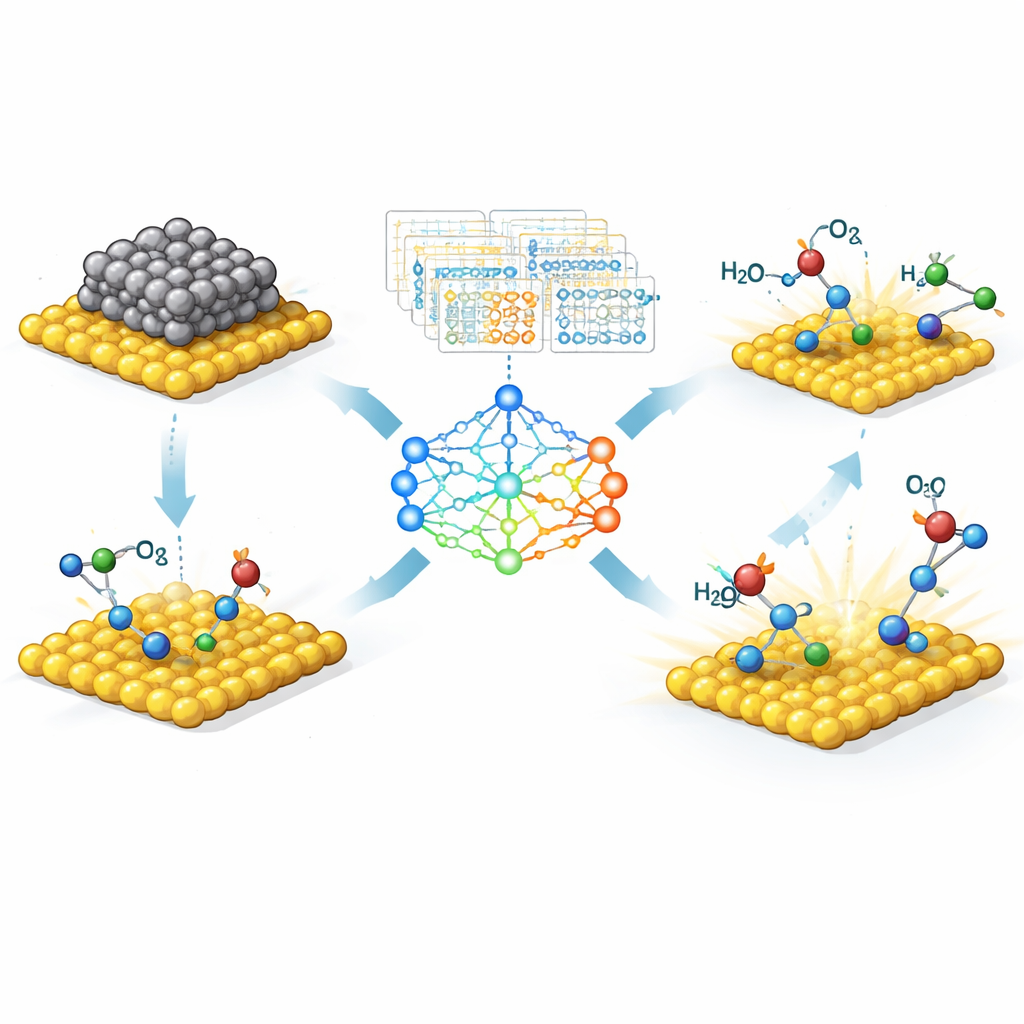
发现强效催化剂背后的隐含规律
文章的核心主题是寻找紧凑的“描述符”,即由基本性质简单组合而成、能可靠预测催化剂性能的量。机器学习能在大量可能性中筛选出为数不多但最为重要的组合,将杂乱的数据转化为清晰的设计规则。例如,金属原子特定轨道的电子数量或金属与载体之间的电荷分布常常能预测关键中间体的吸附强度。在某些情况下,这些规律可以用简短的方程表达,科学家可直接利用这些方程在计算机上筛选数千种潜在的单原子或双原子催化剂,再决定哪些在实验中合成。
确保催化剂能长期耐用
好的催化剂不仅要有高活性,还必须足够耐久。综述描述了机器学习模型如何估计单原子是否会稳定地固定在载体上,或会否聚集成活性较低的颗粒。通过将金属—载体键的强度和金属本身的内聚力与原子扩散和团聚的速率联系起来,作者展示了如何从几个基本数值预测稳定性。这使研究者能够在早期筛除脆弱的设计,集中精力于那些能在高温或腐蚀性溶液等严苛工业条件下生存的材料。
AI 引导的催化剂的下一步方向
展望未来,论文认为机器学习在催化剂设计中发挥全部潜力将依赖三项进展:更好的共享数据库、更智能且更透明的模型,以及与实际工况更紧密的结合。大型、标准化的实验与计算数据集合将使算法学到更普适的规则,而不是零碎的经验技巧。新的“白盒”模型将物理规律与数据科学相结合,既提供准确性又带来洞见,避免难以信任的黑箱预测。最后,通过向模型输入来自试点工厂和运行设备的数据,研究者希望能优化催化剂,不仅适用于理想的实验室测试,而且能在长期、成本有效的工作能量技术中表现良好。
引用: Hu, Z., Wang, Z., Peng, Y. et al. Machine learning-guided design of energy-related catalysts from nanoparticles to single-atom sites. Commun Chem 9, 128 (2026). https://doi.org/10.1038/s42004-026-01967-y
关键词: 机器学习 催化剂, 单原子催化剂, 纳米颗粒催化, 能源转换材料, 数据驱动的材料设计