Clear Sky Science · zh
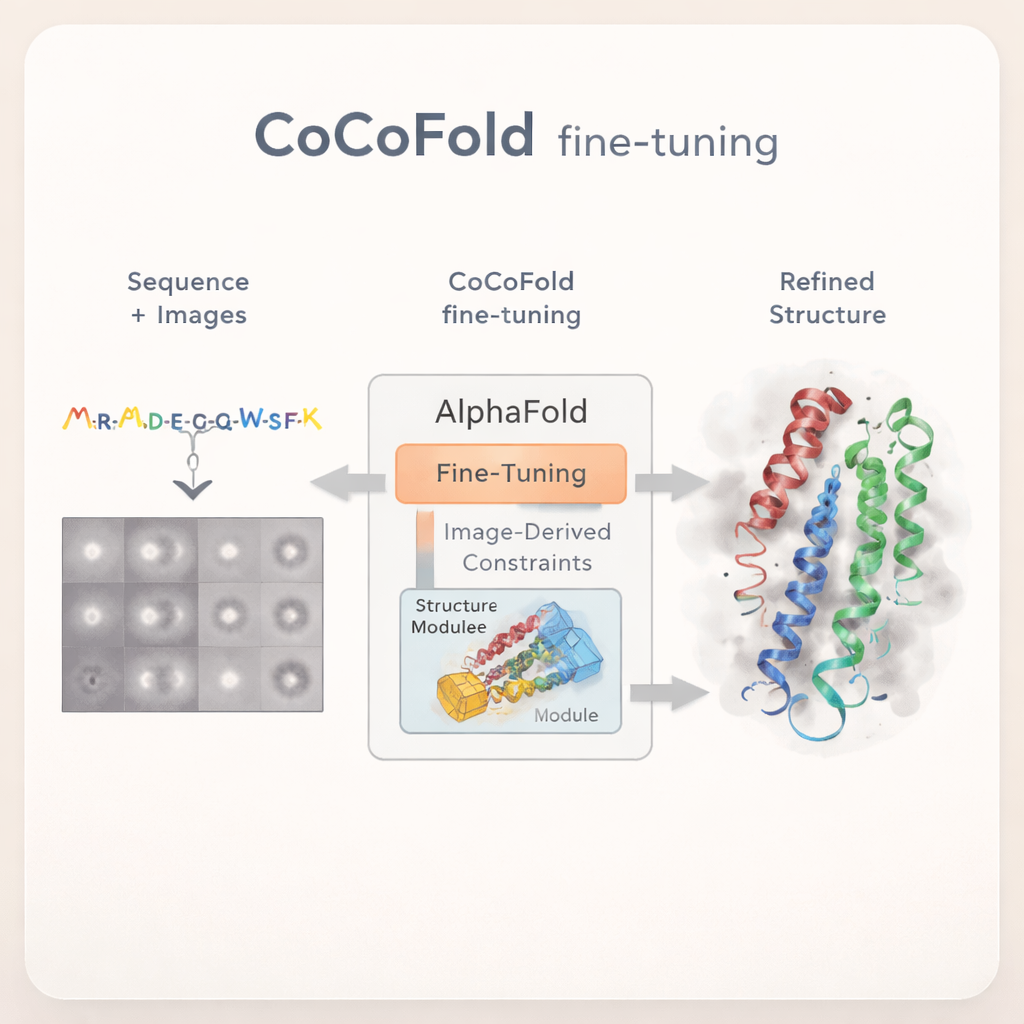
在有限的冷冻电镜观测下微调 AlphaFold
为什么蛋白质形状难以观察
蛋白质是微小的分子机器,驱动着我们体内几乎所有过程,从能量生成到神经信号传递。要理解它们如何工作——以及药物如何调控它们——科学家需要知道它们精确的三维形状。两种强有力的工具为此而生:冷冻电子显微镜(cryo‑EM),通过对冰冻蛋白拍摄大量模糊快照;以及 AlphaFold,一种从氨基酸序列预测蛋白结构的人工智能系统。但在许多实际实验中,cryo‑EM 数据不完整,而 AlphaFold 的预测并不总是与真实情况一致。本文提出了 CoCoFold——一种教会 AlphaFold 直接利用困难的 cryo‑EM 数据并相应改进其预测的方法。
当相机看到的信息太少
cryo‑EM 的工作原理是将蛋白瞬间冷冻,并从多个角度对大量单个颗粒成像,然后将这些图像合成为三维密度图。然而在实际操作中,研究者往往没有足够的优质图像可用。有时蛋白只在高能态下短暂出现,因此捕获到的颗粒非常少;在另一些情况下,蛋白在冰面上偏好某些取向,导致许多观察角度缺失。这两类问题都会产生模糊、不完整的密度图,难以转换为可靠的原子模型。现有软件可以将 AlphaFold 的预测结构拟合到这样的密度图中,但其成功性在很大程度上依赖于一开始就拥有清晰、高分辨率的数据。
教 AlphaFold 从原始图像中学习
CoCoFold 采取了不同的思路:它不依赖完全重建的三维 cryo‑EM 密度图,而是直接使用原始的二维颗粒图像来微调 AlphaFold。该方法从 AlphaFold‑Multimer 的预测出发,保持原网络的大部分参数冻结,保留其关于蛋白折叠的广泛知识。只有最后的构建结构模块允许调整。一个轻量级的“适配器”被加入以将从 cryo‑EM 图像中提取的信息输入到该结构模块中,温和地推动模型朝与实验数据兼容的形状靠拢,同时避免偏离已知蛋白物理规律的极端变化。
将图像转化为结构反馈
为将单个蛋白原子与嘈杂的显微镜图像关联起来,CoCoFold 使用重叠的三维斑点来构建预测结构的平滑、可变形表示,这种表示称为高斯混合。从这一表示出发,它模拟蛋白在与真实实验相同观察方向和成像条件下在显微镜中的样子。然后将这些模拟快照与实际的 cryo‑EM 颗粒在频率域中逐环比较,以评估匹配程度。任何不匹配都会成为反馈信号回流到网络,微调蛋白模型和密度表示。训练完成后,对原子模型进行基于物理的精修步骤,以消除局部几何冲突。
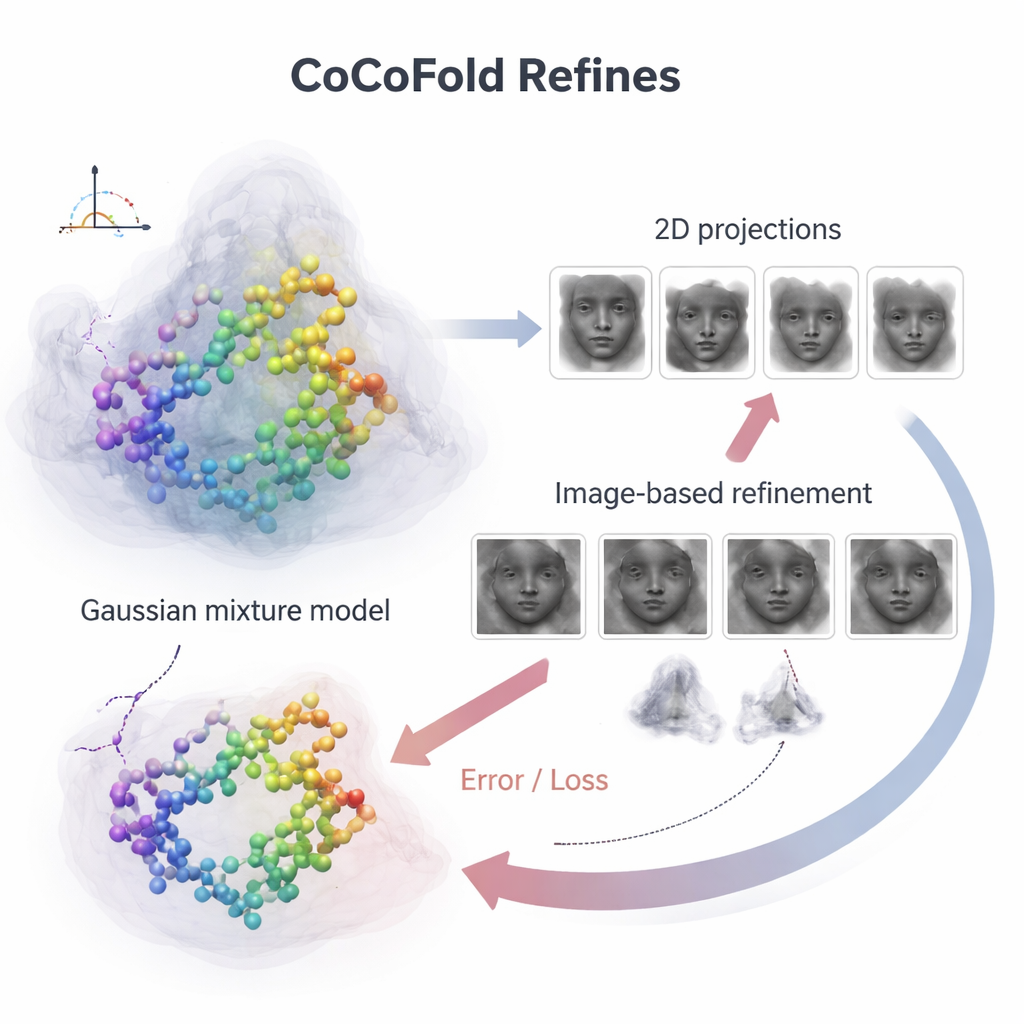
在数据稀少或有偏时保持准确
作者在若干实验和模拟数据集上测试了 CoCoFold,这些数据集旨在模拟 cryo‑EM 的两类主要问题:颗粒过少和观测角度存在大缺口。在这些严苛条件下,标准工具——包括那些依赖重建密度图的其他深度学习方法——往往会错过蛋白的某些区域、错置螺旋或在密度图变模糊时丢失精细结构。相比之下,CoCoFold 始终产生与已知参考结构更为接近和更完整的模型。即便在颗粒数量大幅减少或大量观测方向缺失时,其误差仍保持较小,这表明直接从原始图像学习保留了被基于地图的方法丢弃的重要信息。
这对未来结构生物学的意义
对非专业读者来说,关键的信息是:CoCoFold 像是在强大的 AI 预测与不完美的实验数据之间搭建了一座翻译桥梁。它不再单方面信任 AlphaFold 或 cryo‑EM,而是让二者互相补充,尤其是在实验仅提供部分视角的困难情形中。在样本充足、数据质量高的简单情况下,现有的基于密度图的工具仍然表现优异。但在颗粒稀少或取向缺失——常见于追踪短暂或脆弱蛋白状态时——的情况下,CoCoFold 为从原本会被放弃的信息中恢复可靠的原子模型提供了一条可行路径。
引用: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
关键词: 冷冻电镜, AlphaFold, 蛋白质结构, 深度学习, 结构生物学