Clear Sky Science · zh
从头算确定动态无序固体的相稳定性:Li2C2 中的 C2 旋转无序
为何这种可变的固体重要
许多现代技术依赖于在受热或受压时能够安静改变其内部结构的固体。这些变化称为相变,是固态制冷和更安全电池等概念的核心。本研究考察了一种简单化合物——碳化锂(Li2C2),它随温度升高从整齐有序的形式转变为更加躁动、动态无序的形式。通过在计算机模拟中逐原子观察这一转变,作者展示了微小分子单元的内部“躁动”如何能左右两种晶体结构之间的平衡。
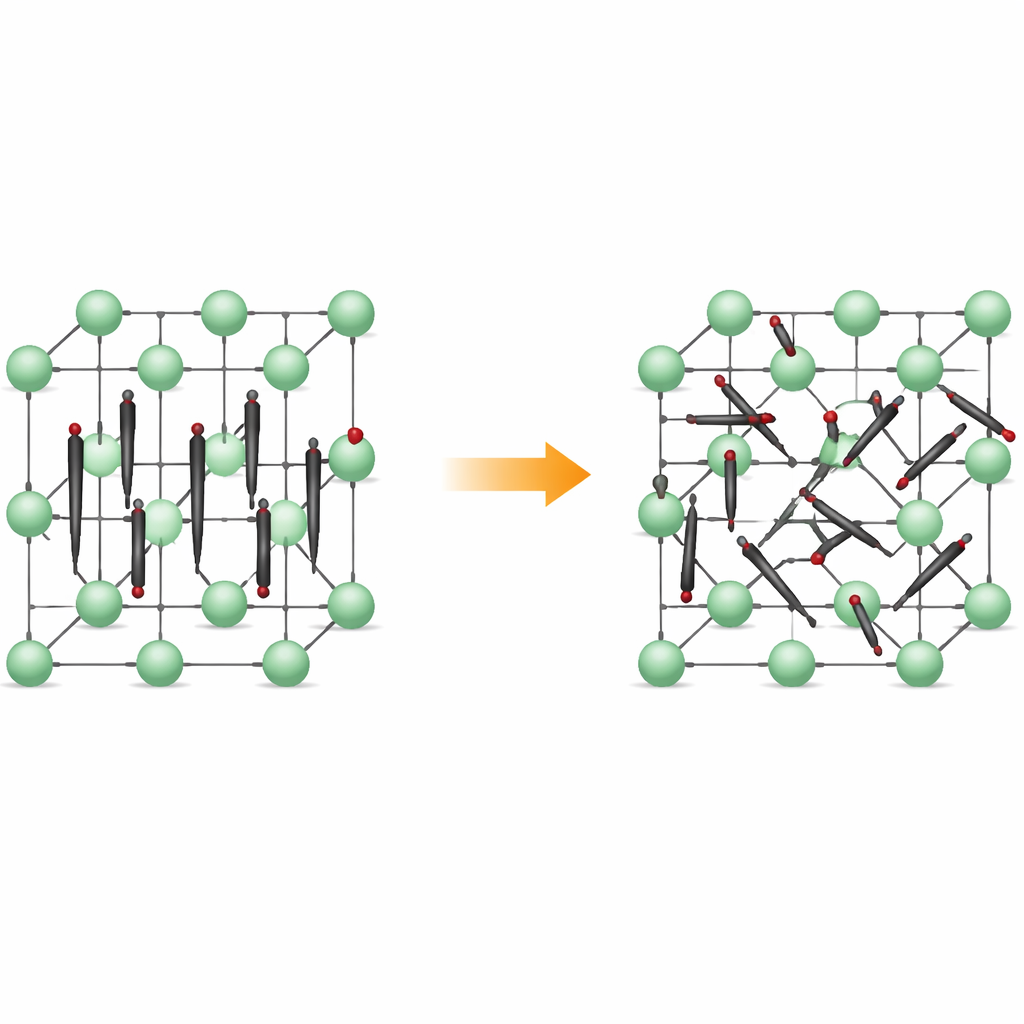
从整齐行列到不安运动
在低温下,Li2C2 形成正交晶:碳原子成对形成小的 C2 二聚体,几乎都指向相同的方向,如同排成一列的火柴棒。锂离子位于其间,构成规整的三维框架。材料受热后转变为立方相,其中二聚体中心的位置仍在晶格上有序,但二聚体自身不再保持固定方向。相反,它们在数个优选取向之间旋转,在对应特定排列的浅能谷中停留。材料仍为固体,但其内部结构变为动态无序。
沿平滑路径追踪变化
要判断在给定温度下哪个相更稳定,必须比较它们的自由能,自由能综合了能量和熵(无序度的度量)。基于固定位置小振动的标准方法在原子大量移动或旋转时会遇到困难。在此,作者使用一种称为应力-应变热力学积分的技术,基于从头算分子动力学。他们构建了一条平滑的形变路径,将模拟胞从低温正交结构连续重塑到高温立方结构。沿该路径,他们在固定温度下进行长时间模拟,测量内部应力对施加应变的响应。对该应力响应积分得到两相之间的自由能差。
通过原子运动看见熵
计算结果显示,在大约 600 K 时低温正交相仍略占优势,而在 650 K 时立方相以每个化学式单位几千分之一电子伏特的优势获胜。对这些结果插值得出相变温度约为 611 K。这低于实验估计,但考虑到自由能差很小,仍属合理范围。立方相的内能实际上更高;使其稳定的是来自 C2 二聚体旋转无序带来的巨大熵增。通过分析每个二聚体的取向如何随着时间丧失对初始方向的记忆,作者表明二聚体在亚皮秒时间尺度上重新定向,模糊了“振动熵”和“构型熵”惯常分类之间的界线。
超越关于固体无序的简单图景
这项工作还强调了常见简化做法的局限性——例如将熵简单地视为围绕固定构型的振动之和加上静态取向的独立计数——在像 Li2C2 这样的材料上会失效。由于二聚体旋转既快又与普通振动强耦合,系统无法被干净地分割为“振动”与“重排”两个独立部分。应力-应变积分方法绕过了这一困难:它直接从微观动力学中提取完整的自由能,无需对熵的划分进行假设。
研究教给我们的东西
通俗地说,该研究显示了固体如何在保持刚性的同时,其内部构建单元变得越来越自由地扭动和转动,以及这种内部自由如何使更无序的结构在热力学上更受青睐。对于 Li2C2 来说,高温立方相并非因为能量更低而被稳定,而是因为它为 C2 二聚体提供了更多的取向和运动方式。通过证明应力-应变热力学积分能够捕捉秩序、能量与熵之间的这种微妙平衡,这项工作为预测其他可能支撑未来制冷装置、电池与智能材料的动态无序固体中的类似相变开辟了道路。
引用: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
关键词: 固态相变, 动态无序, 分子动力学, 碳化锂, 热力学积分