Clear Sky Science · zh
使用遗传数据的混合调优深度学习模型用于乳腺癌诊断
这对患者和家庭的重要性
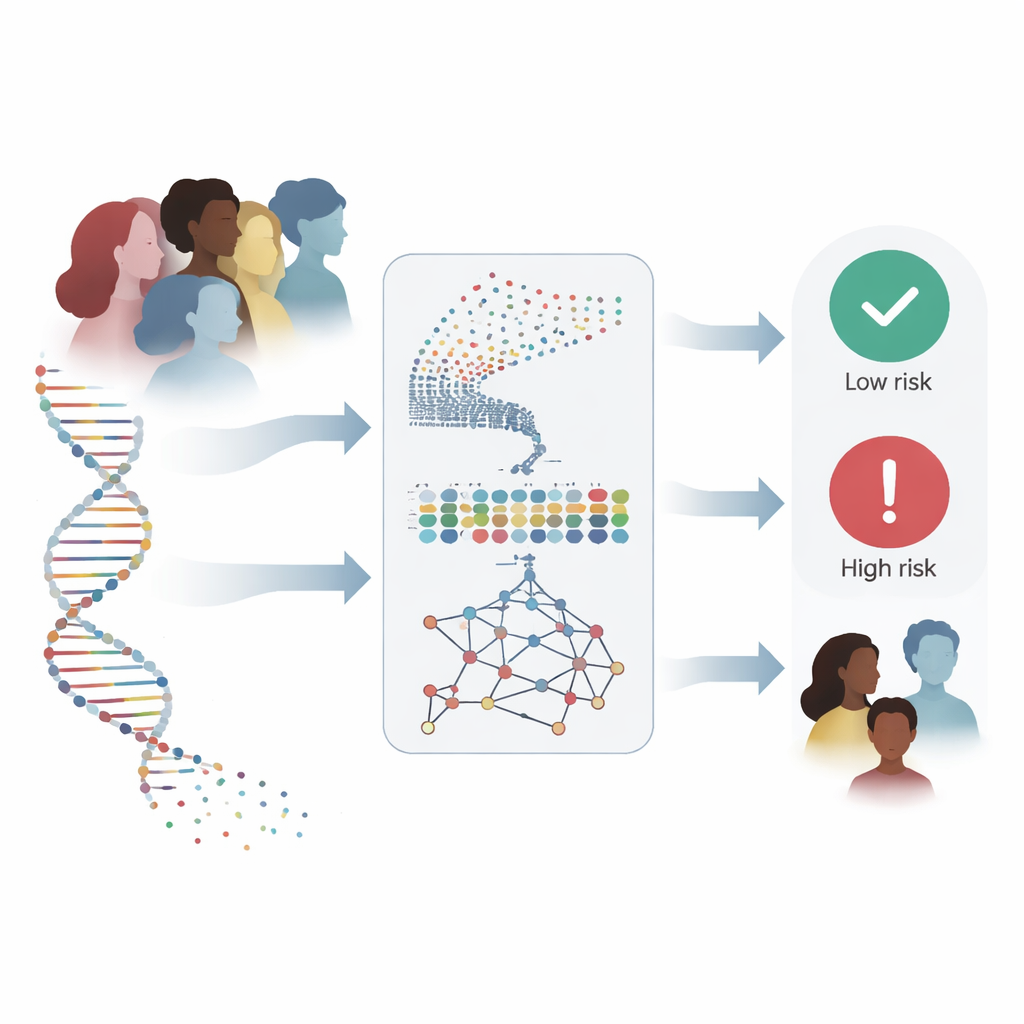
乳腺癌目前已成为全球女性中诊断最常见的癌症,早期发现往往意味着生与死的差别。医生越来越多地可以获得个体的遗传信息,但要把成千上万的基因测量值转化为明确结论极为困难。本文描述了一种新的计算模型,它可以读取这些复杂的遗传模式以识别乳腺癌并高精度地预测结局,可能为临床医生在更早、更可靠的决策上提供强有力的辅助。
从基因到预警信号
每个乳腺肿瘤都携带着一种分子指纹,体现在数千个基因的表达活动中。作者旨在构建一个能够直接读取这种指纹的系统,而不是仅依赖影像或少数已知基因(例如BRCA1和BRCA2)。他们使用了两个癌症基因组学领域最大的公共资源:TCGA乳腺癌队列,包含590个样本中17814个基因的表达数据;以及METABRIC研究,包含1400多名患者的基因组和临床信息。目标很大胆:设计一种方法来处理这股信息洪流,找到最有意义的信号,并在完全独立的患者人群中仍能可靠工作。
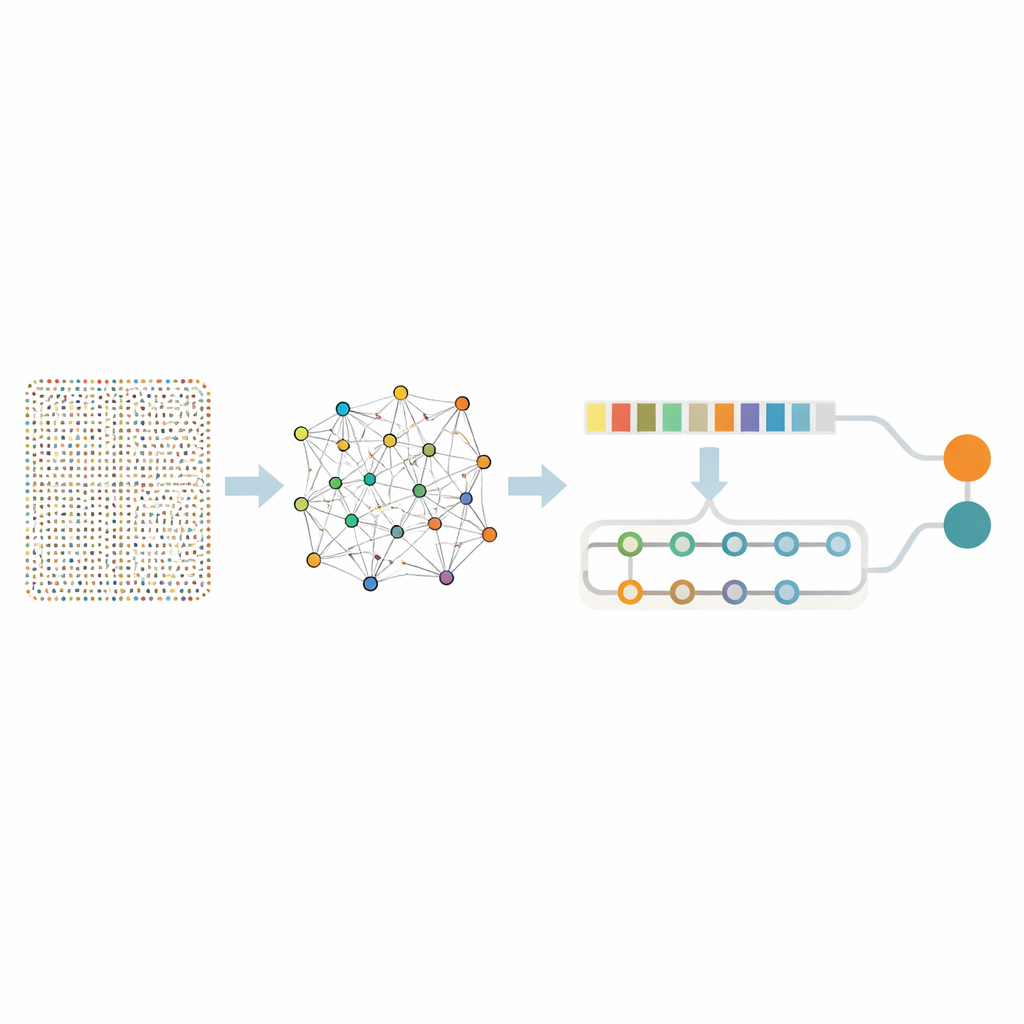
将数千个基因精炼为有用集合
一个学习模式与上下文的双部分神经网络
有了这个聚焦的基因集合,团队构建了一个混合深度学习模型,结合了两类神经网络。一部分为卷积网络,沿着基因列表扫描以捕捉局部模式——即倾向于同时上调或下调的基因簇。第二部分为双向记忆网络,它在同一信息上跟踪长程关系,捕获远端基因在整体谱中如何相互影响。在训练之前,作者对数据进行平衡处理,使癌症与非癌症样本得到公平代表,并加入少量人工噪声,训练模型不要被随机波动误导。
系统在现实数据测试中的表现
在TCGA数据上训练和测试时,混合网络以约97%的准确率正确区分肿瘤与正常样本,并表现出近乎完美的分离能力。重要的是,它优于更简单的深度学习结构和常规机器学习工具,如逻辑回归与支持向量机,即便这些对照方法使用了相同的精心挑选的基因。最强的检验是模型能否在完全不同的数据集上保持性能。应用于在其他医院、使用不同实验方法收集的METABRIC数据时,系统仍保持高性能:在表现最好的运行中,它达到了99.3%的准确率,并正确识别出随后死于乳腺癌的每一位患者——如果该工具用于标记高风险病例,这一特性至关重要。
这可能对未来护理意味着什么
对非专业读者而言,结论是这项研究提供了一个智能的遗传数据筛选和解读工具,能够在大规模患者群体中以显著一致性识别乳腺癌及相关风险。通过将周到的基因选择策略与双分支神经网络相结合,作者展示了计算机可以从庞大的基因数据集中提取临床有意义的信号,而且这种能力不仅适用于单一研究,也可跨独立队列复现。尽管还需要在多样化人群中进一步验证该方法并详细解释其决策过程,但该方法指向了一个未来:简单的血液或组织样本可输入此类模型,帮助医生更早发现肿瘤并更精确地定制治疗。
引用: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
关键词: 乳腺癌基因组学, 深度学习诊断, 基因表达生物标志物, 早期癌症检测, 临床决策支持