Clear Sky Science · zh
DPAS:用于通过单类学习识别致病肽的疾病相关肽异常评分
为什么微小的蛋白片段对健康很重要
肽——短链蛋白片段——已成为现代医学中的新宠。它们可以在体内充当精确的信使,并且日益被用作药物和疾病标记。然而,要确定哪些肽真正与疾病相关通常依赖于明确的“疾病”与“非疾病”示例,而生物学很少提供这样的清晰对照。本研究提出了一种新方法,利用仅有的已知疾病相关肽来识别潜在有害肽,提供了一条更快速、偏倚更小的途径来发现未来的诊断手段和治疗靶点。
找到“非疾病”组的难题
传统计算模型通过比较两个类别来学习:已知与疾病相关的阳性样本和被认为无害的阴性样本。在肽研究中,第二组就是问题所在。许多肽根本未被测试,因此将它们标记为“非疾病”可能具有误导性并引入偏差。以往关于抗癌或抗炎肽的研究虽取得令人瞩目的准确率,但常依赖人工构建或推测的负样本数据集。因此,这些模型在面对罕见信号或与训练数据不相似的新型疾病肽时可能表现不佳。
从已知中学习,而非从猜测出发
作者采取了不同路径:他们不将问题强行变成双侧对比,而是将疾病相关肽视为一个内在一致的群体,问道:“这个群体的细节特征是什么?”他们从一个专门的癌症相关数据库中收集了超过76万条突变的人类肽,并用一组丰富的特征来描述每条肽。这些特征包括各氨基酸的出现频率、氨基酸对的排列方式、体积与亲水性等基本理化性质,以及称为基序的短重复序列模式。随后使用主成分分析将这种高维描述压缩为更易处理的形式,同时保留主要的变异来源。
用单类模型发现异常肽
在得到压缩后的特征空间后,团队训练了三种“单类”模型——旨在学习单一群体形状并标记不符合者的算法。他们测试了一类支持向量机、隔离森林以及一种称为自编码器的神经网络。自编码器学习将每个肽的特征压缩到狭窄的内部表示再重建;属于已学疾病模式的肽能被准确重建,而异常肽会产生更高的重建误差。将各方法的归一化异常分数进行比较表明,自编码器产生了最紧密的典型肽簇并在内群与异常值之间实现了最清晰的分离。通过在重建误差的95百分位左右设定阈值,模型将大多数肽归类为可能与疾病相关,同时稳定地标记出一小部分为非典型肽。
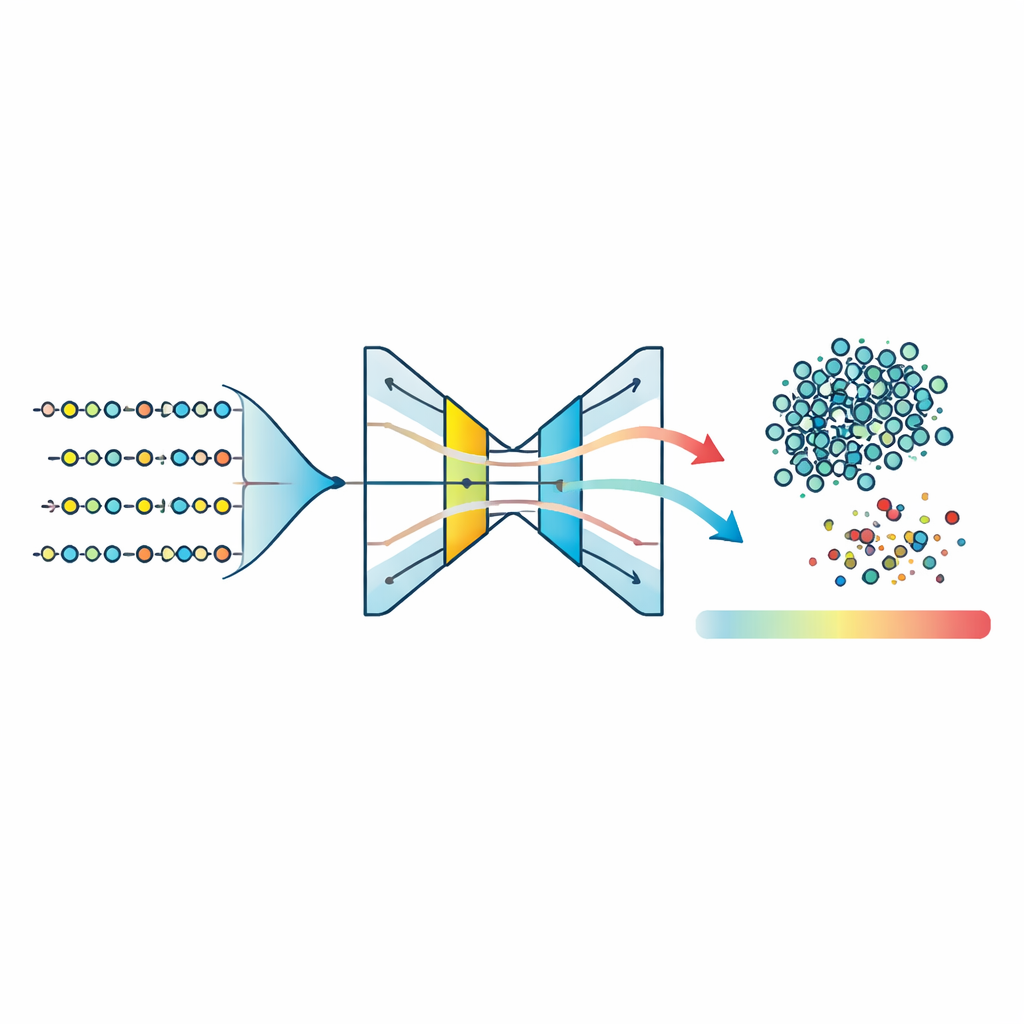
把复杂评分变成单一有意义的数值
为了让结果在生物学上更易解释,作者引入了疾病肽异常评分(DPAS)。该评分融合了两类成分:自编码器认为肽有多不寻常(其归一化重建误差)以及其特征在预测中贡献的强弱,这由一种流行的解释方法SHAP来衡量。实际上,基序和特定的理化性质表现出特别的信息量。DPAS将这些信号组合,使得既在结构上异常又由生物学上有意义特征支持的肽获得更高排名。然后对得分最高的肽使用基序搜索工具,连接到已知的功能性特征,例如磷酸化位点、金属结合区域以及其他常见于信号传导和酶调控的调节模式。
这对未来诊断和药物意味着什么
通俗地说,这项工作提供了一种更智能的筛选方法,可以在不假定哪些肽绝对无害的前提下找到可疑肽。通过仅从已证实的疾病相关示例中学习,然后用DPAS对新候选肽排序,研究人员可以优先选择一小批在生物学上可信且便于实验验证的肽。许多高分候选肽包含已知的功能性基序,这强化了它们可能在疾病过程中发挥作用的想法。尽管该方法仍依赖某些假设且缺乏经实验验证的“安全”肽以供全面验证,但它为肽类生物标志物发现提供了更现实和透明的基础,并可扩展至其他缺乏可靠阴性样本的生物数据类型。
引用: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
关键词: 疾病相关肽, 异常检测, 自编码器, 生物标志物发现, 单类学习