Clear Sky Science · zh
苏丹大肠埃希氏菌中典型与新型耐药相关错义突变的结构建模与对接分析
这对日常健康意味着什么
抗生素耐药性感染已不再是罕见的医学奇观;它们正越来越威胁着泌尿系感染、外科手术和重症监护等常规治疗。该研究聚焦于来自苏丹的大肠埃希氏菌,提出一个具体问题:细菌蛋白中的微小基因变化如何重塑常用抗生素的作用?研究者采用基于计算的结构建模替代昂贵的实验室方法,揭示了标准检测和全球数据库可能遗漏的隐性耐药模式——尤其是在耐药上升最快、资源有限的地区。 
探查细菌的“工具箱”
研究者关注的是“错义”突变——将蛋白中一个氨基酸替换为另一个的单个碱基变化。他们分析了55株来自苏丹的大肠埃希氏菌的全基因组序列,聚焦于作为主要抗生素类别直接靶标的细菌蛋白,包括氟q喹诺酮类、大环内酯类和利福平类等。这些靶点包括改变DNA拓扑的酶(旋转酶和拓扑异构酶IV)、负责蛋白合成的核糖体,以及RNA聚合酶。在这些蛋白中发现的71处突变中,有19处被多个预测工具标记为可能损害蛋白功能,显著的是,大多数这些突变似乎是尚未被全球耐药数据库收录的新变异。
熟悉靶点中的新问题区域
一些最重要的变化聚集在核糖体蛋白L22,该蛋白参与形成新合成蛋白质穿出核糖体的通道。该区域同时也是大环内酯类抗生素(如红霉素)的对接位点。研究发现了一组此前未报道的L22突变,且许多突变出现在同一菌株内,位置正好位于该通道及与核糖体RNA接触的点上。计算分析表明,其中若干改变使局部结构不稳定或更具柔性,可能重塑通道形状,从而使大环内酯分子与其结合的位置不再契合。同时,典型的“经典”耐药突变出现在DNA加工蛋白ParC和ParE以及RNA聚合酶中,证实苏丹菌株既具有一些全球性耐药特征,也带有其本地化的变体。
构象变化如何削弱抗生素结合
研究团队不仅列出序列差异,还探讨这些突变如何改变抗生素与其靶标之间的三维配合。通过分子对接模拟,他们比较了不同药物与正常及突变蛋白的结合方式。对于拓扑异构酶IV蛋白ParC,位于药物接触位点附近的关键突变显著削弱了氟喹诺酮类药物托伐氟沙星的预测结合,反映出在酶–DNA–药物界面处结合变得更为松散。在相关的ParE蛋白中,突变使诺氟沙星(novobiocin)的结合略有降低。相反,旋转酶GyrA中的一处新突变似乎使酶结构不稳定,但并未明显改变氟喹诺酮类药物莫西氟沙星的结合紧密度,这表明耐药有时可以通过微妙扰乱酶的性能而产生,而不仅仅是通过将药物排斥出结合位点。
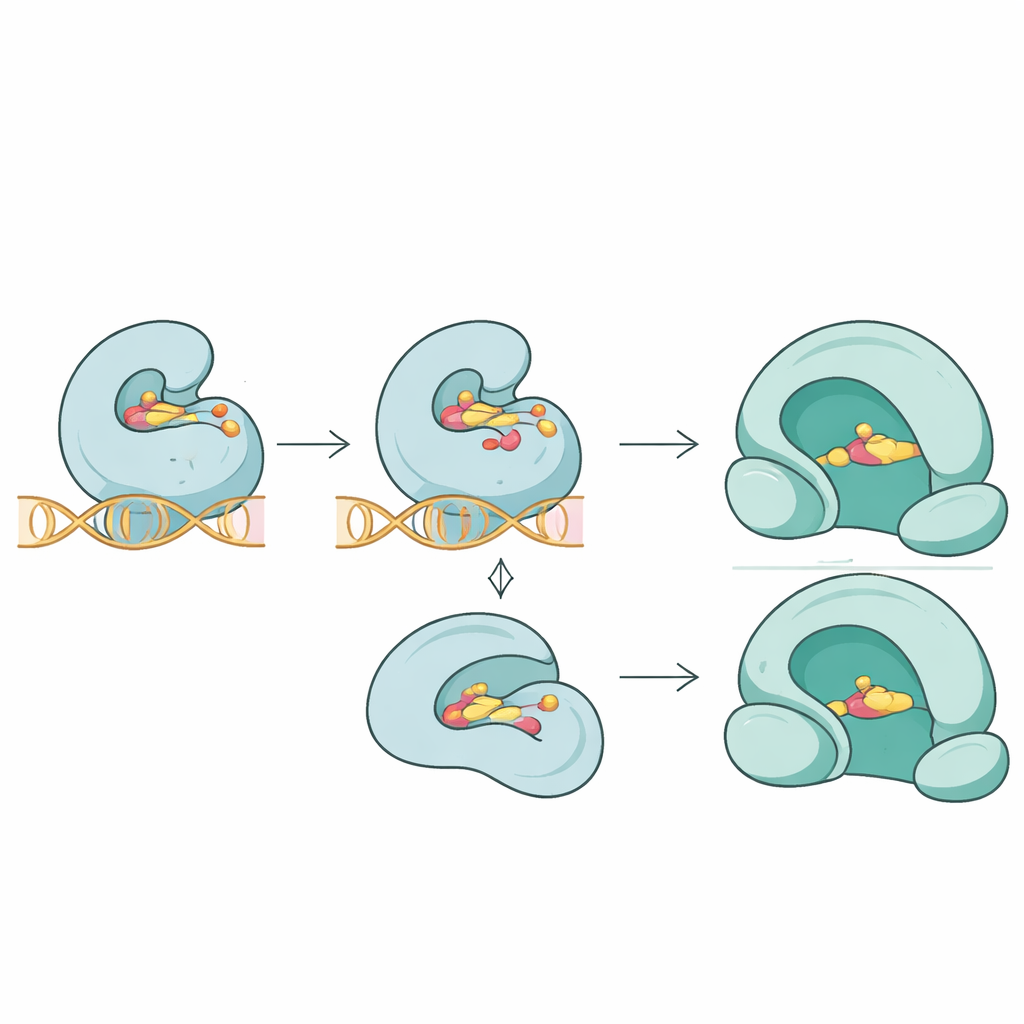
对不同药物的混合效应
并非所有突变的影响都相同。RNA聚合酶RpoB中的经典利福平耐药性改变对一种结构上不同、靶向相邻位点的新抑制剂结合影响甚微,这意味着未来药物可以被设计成绕过现有的耐药模式。对于核糖体蛋白L22,与红霉素的对接研究显示出一系列结果:有些突变削弱了结合,有些影响不大,甚至有一处略微改善了预测结合。这些结果强调耐药很少是全有或全无的;每个突变都会以不同方式推动蛋白稳定性、柔性和药物结合的变化,而对治疗的总体影响取决于这些变化在活细胞中如何组合。
对患者与监测工作的意义
从通俗角度看,关键信息是像苏丹这样的地区的细菌通过已知与未知的多条路径演化出耐药性。已知路径包含国际项目已追踪的经典突变,但本研究表明还有许多在当地富集的额外突变,可能以更微妙的方式削弱抗生素。通过将这些变化映射到详细的蛋白结构上,作者提供了一份应在实验室中验证并纳入地区诊断面板的突变名单。在实际层面,他们的工作表明,智能的计算建模可以帮助实验能力有限的国家更好地监测新兴耐药,最终支持更可靠的治疗选择,并为设计能领先细菌进化一步的新药提供启发。
引用: Sage, E.E., Ibrahim, S.A.E., Firdaus-Raih, M. et al. Structural modeling and docking analysis of canonical and novel resistance-associated missense mutations in Sudanese Escherichia coli. Sci Rep 16, 8995 (2026). https://doi.org/10.1038/s41598-026-39491-7
关键词: 抗菌素耐药性, 大肠埃希氏菌, 错义突变, 结构生物信息学, 苏丹