Clear Sky Science · zh
CiCLoDS:用于单细胞空间转录组学的联合细胞聚类与基因选择
在细胞之城中寻找“街区”
现代显微技术现在可以在保留每个细胞在组织中原位位置的同时,读取成千上万个细胞中哪些基因处于活跃状态。这种“空间转录组学”革命就像把模糊的城市地图变成了每栋房屋的街景视图。但问题是:这些地图对每个细胞测量的基因数量高达数千,远超科学家在后续实验中能轻松解释或负担的范围。本研究提出了CiCLoDS,一种新方法,能够发现有意义的细胞邻里,同时挑选出一小组可解释的基因来定义这些邻里。
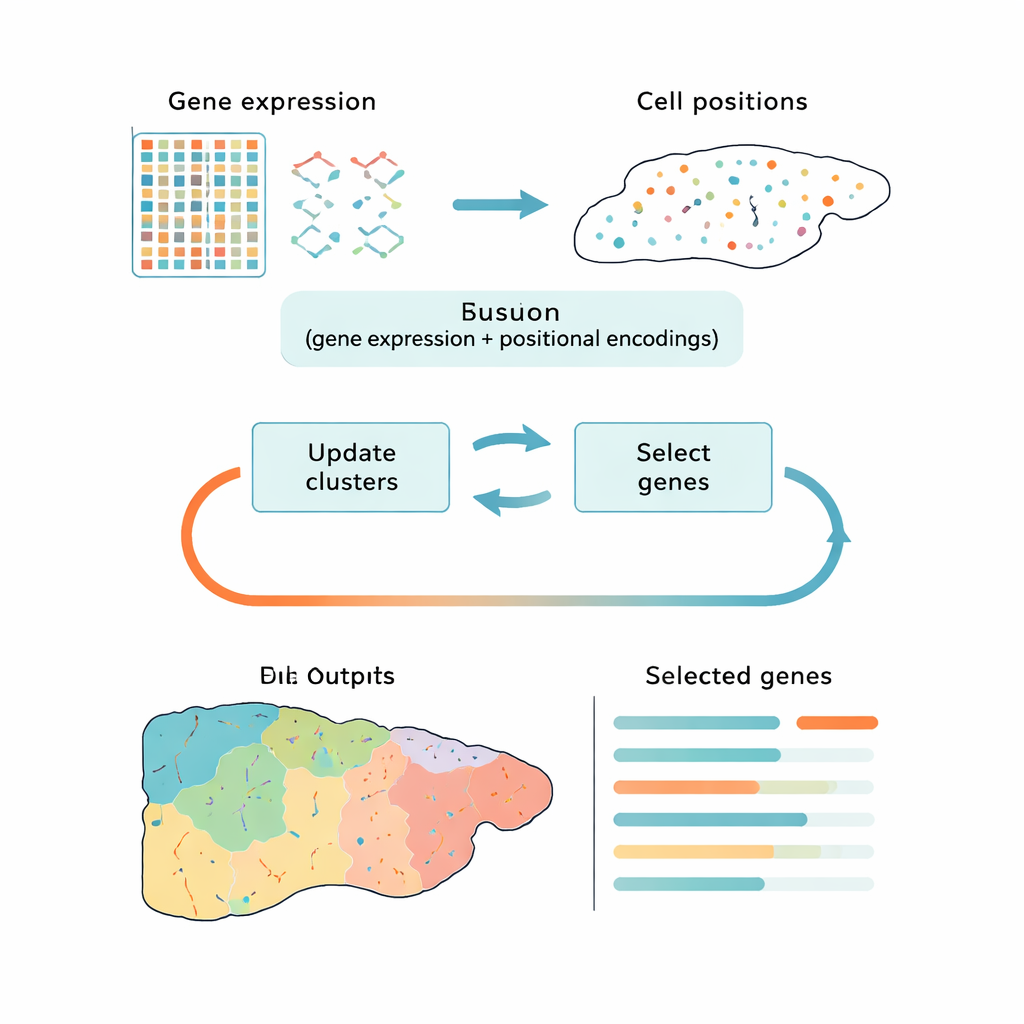
更聪明的数据压缩方法
目前大多数工具以两个相互独立的步骤来处理这个挑战:先将数据降到更简单的形式,然后再把细胞分组。诸如主成分分析(PCA)之类的流行方法虽能保留总体变异,但可能侧重于技术噪音或通用的细胞周期信号,而非真正重要的生物学差异。另一些方法使用深度学习来发现模式,但其表现为黑箱,不能明确指出哪些基因最为关键。CiCLoDS走的是不同路线。它将基因选择和聚类视为在用户定义的“预算”限制下的一个联合问题——即在可保留的有限基因数内,哪些基因最能解释细胞如何基于其基因活性以及(在有时空信息时)其在组织中的位置而分成不同群体?
从数学到真实组织的地图
作者将一类数学上透明的技术——子空间聚类(subspace clustering)——适配到空间转录组学的现实场景中,后者的数据集规模可超过一百万个细胞。CiCLoDS基于简单的细胞×基因表工作,既将细胞归入簇,又为每个基因打分,评估其在区分这些簇时的作用。当需要时,它还能通过加入描述每个细胞在组织中位置的位置信息“编码”来融入空间信息,而不改变核心优化过程。在由高分辨率成像平台生成的大型小鼠肝脏与人结肠数据集中,CiCLoDS可在普通计算机上几分钟内运行,生成紧凑的基因面板——通常为几十到几百个基因——却仍能捕捉原始数据的丰富结构。
揭示隐秘区域与血管
将CiCLoDS应用于小鼠肝脏时,研究团队检验了该方法能否重建已知的“分区”模式——即从肝小叶一侧到另一侧的肝细胞功能逐渐变化。与PCA和一种领先的基因选择工具geneBasis相比,CiCLoDS产生了更清晰的空间分区、边界更锐利且误分配区域显著更少,这在与参考地图一致性的定量指标中得到了体现。令人注目的是,在允许使用更多基因的情况下,CiCLoDS重新发现了类似门静脉区和中心静脉区的肝细胞群,这些群组与专家定义的参考簇高度吻合——尽管方法并未被告知关键标志基因AXIN2,也没有被直接提供空间坐标。当加入空间编码时,CiCLoDS还学得了富含细胞表面与血管相关功能的基因面板,并能准确区分真实血管与成像伪影——这是更简单的方法要么做不到,要么只能通过更多临时性调整才能实现的。
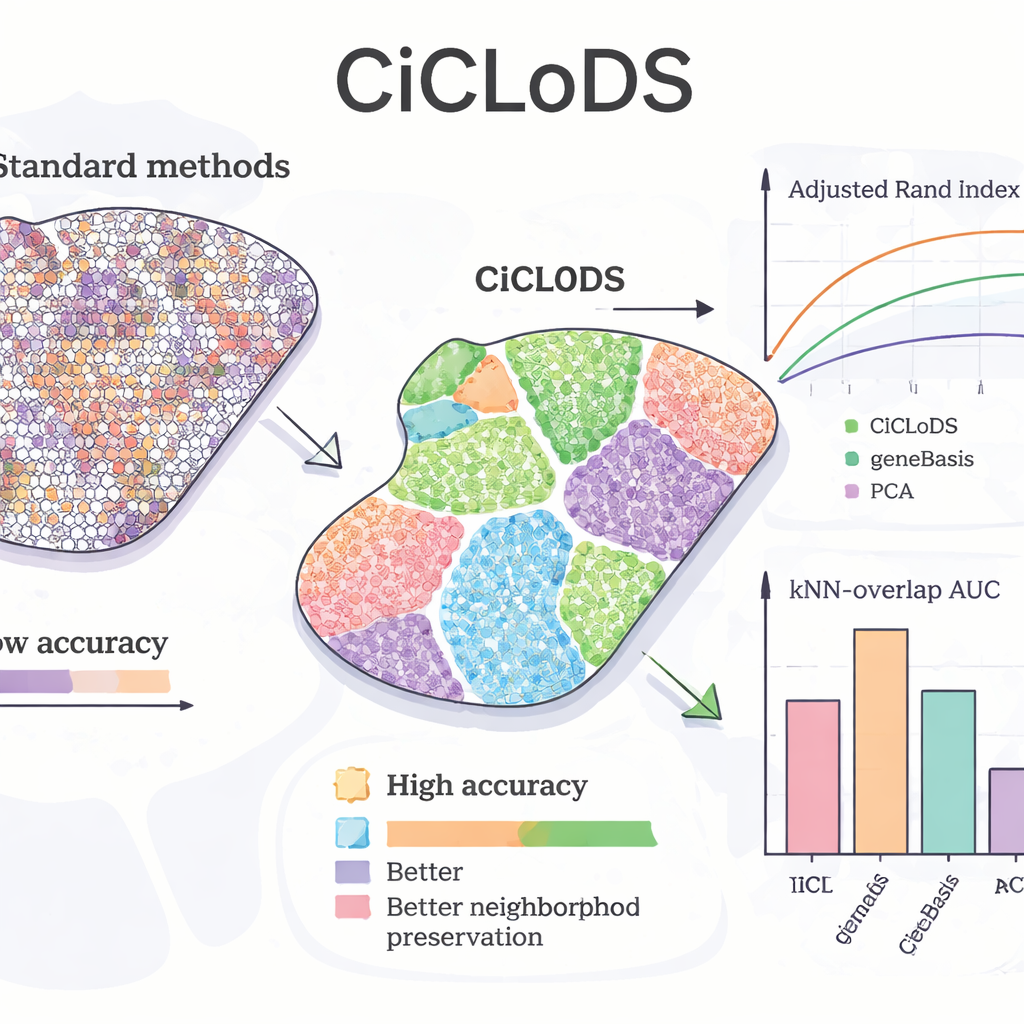
跨脑组织的泛化能力与提升其他方法
为检验CiCLoDS在非常不同的组织与个体间的稳健性,作者分析了来自三位供体的人类背外侧前额皮质样本。在这些样本中,CiCLoDS的表现与专门的空间方法(如BayesCafe和BayesSpace)相当或更优,尤其是在其他工具表现困难的一个样本上。本研究还凸显了一种“混合”用法:先运行CiCLoDS以获得稳定簇,然后将其结果作为初始输入提供给BayesSpace。这种热启动策略提高了整体准确性,并产生了与专家注释最为一致的大脑层状模式,表明CiCLoDS既能独立使用,也能使下游的概率模型更可靠。
这对生物学与医学为何重要
对非专业读者而言,关键结论是:CiCLoDS能将压倒性的细胞地图转化为简洁且具生物学意义的汇总结果。研究者不再需要面对成千上万条嘈杂的测量值,而是得到一份可管理的基因清单和反映真实组织结构的清晰空间簇——例如肝脏的代谢分区、血管及其微环境,以及大脑的层状结构。由于基因预算由用户控制且计算开销小,CiCLoDS可用于设计针对性的基因面板以供未来实验、指导复杂空间数据集的解读,并为更复杂的建模提供稳健的起点。在数据收集已不再是瓶颈、理解成为主要挑战的时代,像CiCLoDS这样的工具有望让高维组织地图既实用又富有洞见。
引用: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
关键词: 空间转录组学, 细胞聚类, 基因面板选择, 组织结构, 单细胞分析