Clear Sky Science · zh
通过原位多化合物合成与原生质谱优化凝集素‑3 结合剂
这对未来药物意味着什么
许多现代药物通过与体内蛋白结合发挥作用,但寻找能牢固且有选择性地贴合特定位点的小分子通常既缓慢又昂贵且令人沮丧。这项研究引入了一种更快的方法,可以在靶蛋白存在的条件下直接微调这些分子,然后用高度灵敏的“称重”技术读取胜出者。作者在与癌症相关的蛋白凝集素‑3 上展示了他们的方法,最终得到一个有前景的类药候选分子,其结合强度可与现有一些最佳化合物相媲美,但结合在蛋白表面一个意外的口袋中。

重新思考我们寻找更好药物先导的方法
传统的药物优化更像是一场昂贵的猜测游戏。化学家对起始化合物逐步改造,测试每一个版本,并希望改善其与靶蛋白的结合能力。但蛋白表面有柔性、水分子会干扰,结合事件本身也可能重塑蛋白,使得计算预测不可靠。即便拥有高分辨率结构,也不能保证建议的修改会有帮助。现有的“靶向引导”方法试图让蛋白从一组构件中自行选择配对,但这些方法仍依赖复杂的分析和间接信号来推断哪个化合物真正结合得最好。
让蛋白选择,然后称出胜者
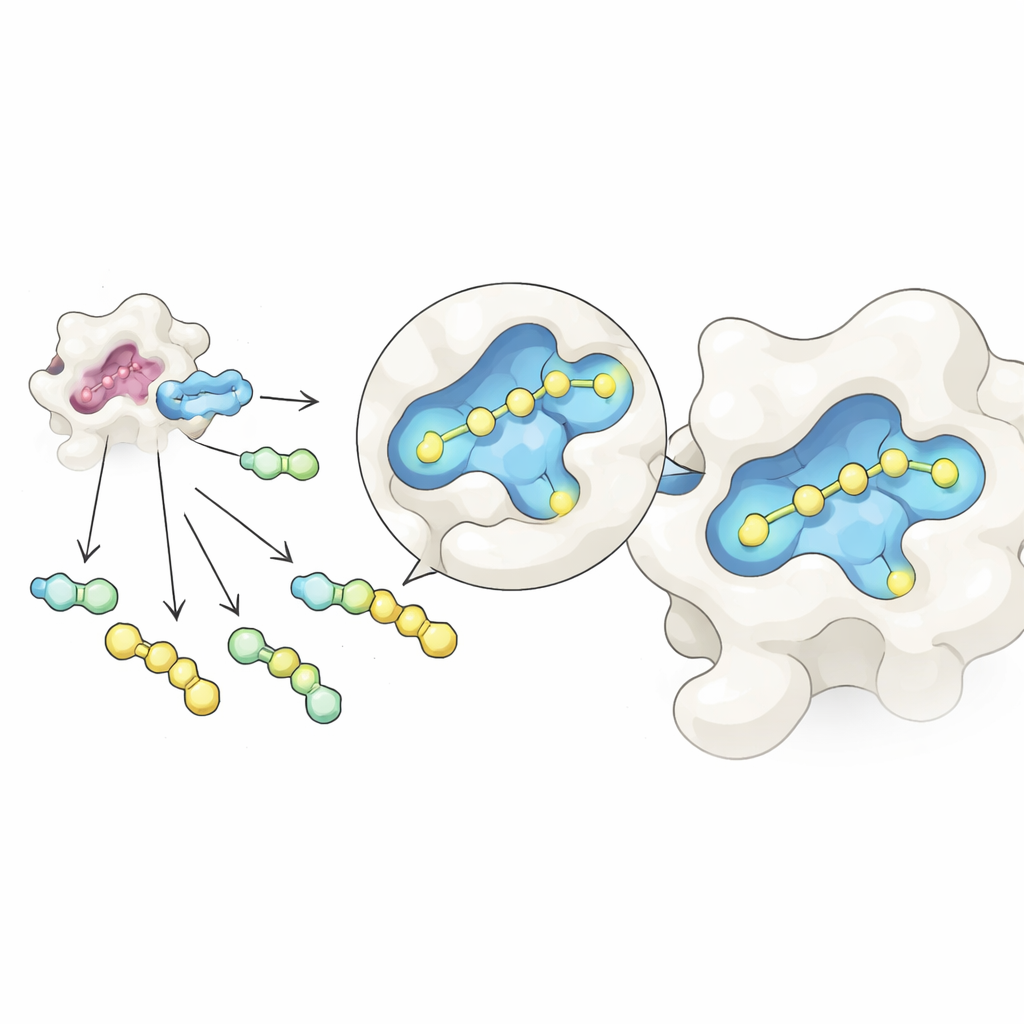
研究人员将两种思路结合到一个简化的工作流程中。首先,他们使用一种可逆化学反应,将一个常见的糖类核心与多种不同的侧基在同一试管内连接,形成一组相关分子的混合物。通过精心调整起始物比例,所得产物在简单的浓度规则下达到平衡,这有助于在原始反应活性不同的情况下均衡各组分的数量。其次,他们将该混合物暴露于凝集素‑3,并用原生质谱进行检测——这是一种在温和、类似水的溶液中保持蛋白‑分子配对完好的质谱方法。由于每个候选分子具有不同的质量,仪器可以直接检测哪些分子真实地与蛋白结合,无需任何标记或参照标识。
从拥挤的混合物中找出突出结合者
利用该配置,团队通过将不同侧基连接到受已知抑制剂 GB1107 启发的糖核心上,创建了数十种凝集素‑3 结合物。他们将 35 种不同的肼酰胺片段分成可管理的小组,在原位形成所有组合,然后加入凝集素‑3。原生质谱突出了那些最常与蛋白伴随出现的化合物,将其标记为主要命中物。一项后续的热稳定性测试(测量化合物在加热时对蛋白的稳定作用)筛除了由气相测量特性引起的假阳性。最终剩下三种领先候选物,详细的热力学结合测定显示其中一种,称为 GalAldBZ20,对凝集素‑3 的结合尤其紧密,达到亚微摩尔量级。
发现隐藏口袋并增强结合
当团队观察 GalAldBZ20 在凝集素‑3 表面的停靠方式时,出现了下一个惊喜。大多数已知结合物使用靠近糖结合位点的“α”口袋,但结构学方法和计算模拟表明 GalAldBZ20 更偏好相邻的“β”口袋。X 射线晶体学给出暗示,溶液中的核磁共振显示该口袋附近存在多种局部构象,分子动力学模拟则支持这样一种模型:分子上的含硝基芳环嵌入到 β 位点。研究人员推断可以通过重新设计糖与硝基环之间的连接子来更牢固地固定这种取向,以促进新的极性接触并降低柔性。
将巧妙的筛选变为有力的候选物
基于这一见解,团队合成了一小组更刚性的后续分子,保留相同的糖和硝基芳环,但改变它们之间的连接方式。其中一个版本——一种 N‑半乳糖苷(化合物 5)脱颖而出:其与凝集素‑3 的结合力约比原始命中物强十倍,达到与 GB1107 相当的结合强度,但仍优先结合 β 口袋。一份超高分辨率的晶体结构显示硝基芳环嵌入该口袋的清晰电子密度,并由若干氢键和与关键氨基酸的阳离子‑π 相互作用支撑。当去除硝基或用简单的甲基取代时,结合明显减弱,强调了该基团的重要性。因为相关蛋白凝集素‑1 缺乏这个 β 口袋,新化合物最终可能提供更好的选择性,这是药物设计中非常宝贵的特性。
这对未来药物发现的意义
通俗地说,这项工作表明你可以将许多相关分子混合在一起,让与疾病相关的蛋白“挑选”其偏好者,然后直接称量这些蛋白‑分子配对,看哪些结合得最好。应用于凝集素‑3,这一策略出人意料地发现并强化了对一个较少探索口袋的结合,得到一个可与一些最佳现有抑制剂媲美的化合物,并可能作为新抗癌药物的先导。更广泛地讲,将原位化学与原生质谱相结合,为针对具有多个潜在结合位点的蛋白优化药物先导提供了一条通用捷径,可能在药物发现早期节省时间、材料和精力。
引用: Hoshi, K., Konuma, T., Taguchi, R. et al. Optimization of galectin-3 binding agents by in situ multiple compound synthesis and native mass spectrometry. Sci Rep 16, 8453 (2026). https://doi.org/10.1038/s41598-026-38570-z
关键词: 凝集素‑3 抑制剂, 原生质谱, 基于片段的药物发现, 靶向引导合成, 癌症药物候选物