Clear Sky Science · zh
基于密度泛函理论的菲咯啉-硝基苯酚配合物的前线轨道与非线性光学特性研究
光、分子与未来技术
日常技术——从智能手机屏幕到高速互联网——都依赖于能够精确控制光与电荷的材料。本研究考察了一对由两种常见有机分子(1,10‑菲咯啉与对硝基苯酚)通过氢键结合并发生电荷共享的微小体系。通过弄清这种“电荷共享伙伴关系”如何形成以及它对光的响应方式,科研人员希望设计出更好的传感器、光学开关和下一代光子器件的组件。
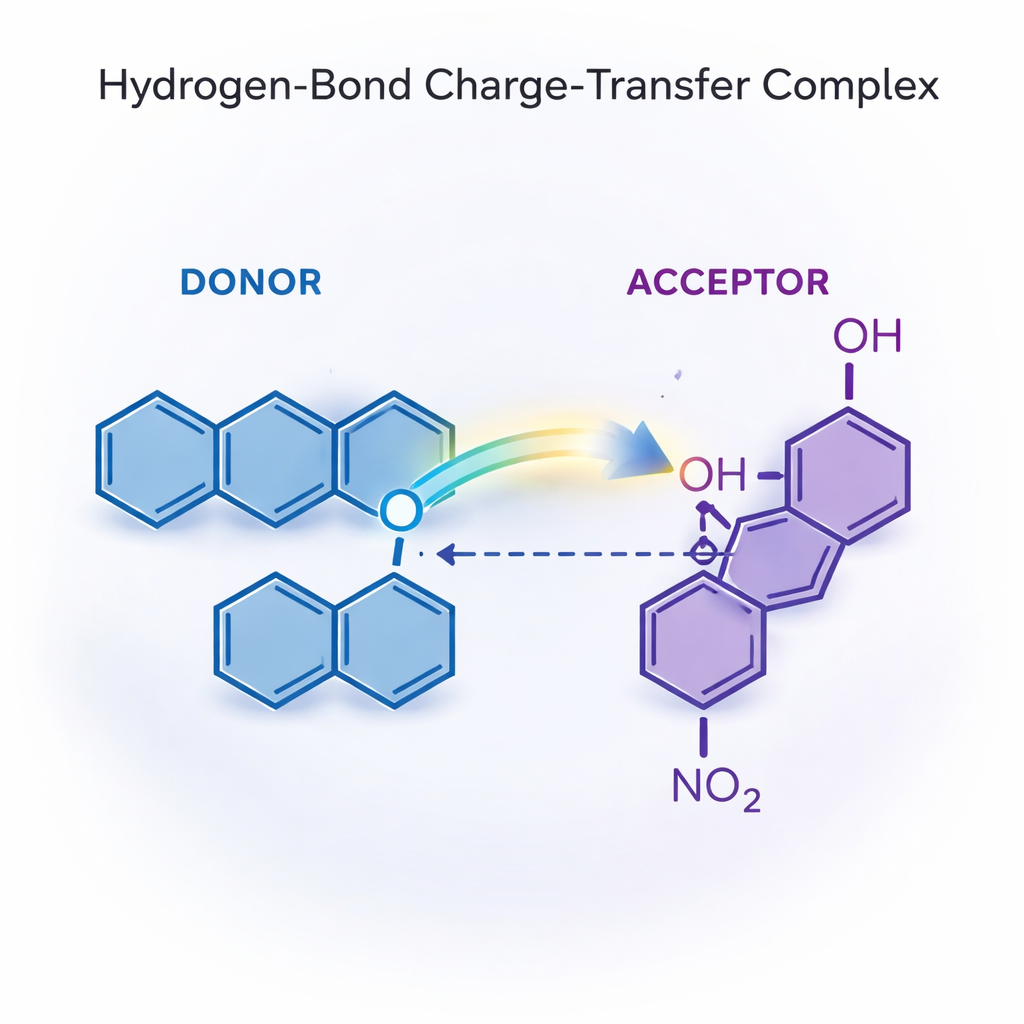
由氢键构建的分子伙伴关系
该工作聚焦于一种特殊的结合类型,称为氢键电荷转移配合物。在这里,一个分子作为电子供体,另一个作为电子受体,氢键像桥梁一样连接两者。作者表明,当1,10‑菲咯啉与对硝基苯酚接近时,对硝基苯酚的酸性氢会向菲咯啉的氮原子偏移,形成强且具方向性的氢键并出现部分质子转移,从而促进电子从一方向另一方移动。结果是一个紧密结合的配对,其结构明显不同于各自分子的孤立结构。
用理论与谱学窥探结构
为揭示该配合物的构造,研究人员将多种实验技术与强大的量子化学计算(即密度泛函理论)相结合。他们模拟原子最优排布,验证预测结构的稳定性,并检查表明强氢键存在的关键键长与键角。红外光谱用于追踪配合物形成时特定振动模式的位移,而核磁共振(NMR)显示氢与碳原子局部电子环境的变化。上述测量共同证实了一个由氢键稳定的真正电荷转移配合物的形成,并且质子在很大程度上已从对硝基苯酚转移到菲咯啉。
电子如何移动与光如何被吸收
研究组接着探讨这种配对如何改变体系的吸光与电荷迁移行为。通过测量与计算的紫外‑可见(UV–Vis)谱,他们识别出一个特征性的电荷转移带:只有在两分子形成配合物时才出现的宽吸收峰。前线轨道分析——观察最高占据与最低未占据的电子态——表明被光激发的电子有效地沿氢键从一个分子片段转移到另一个片段。前线轨道间的能隙显示该配合物电子上相对稳定,但主要在紫外光区有响应,这对于紫外响应材料是有用的特性。
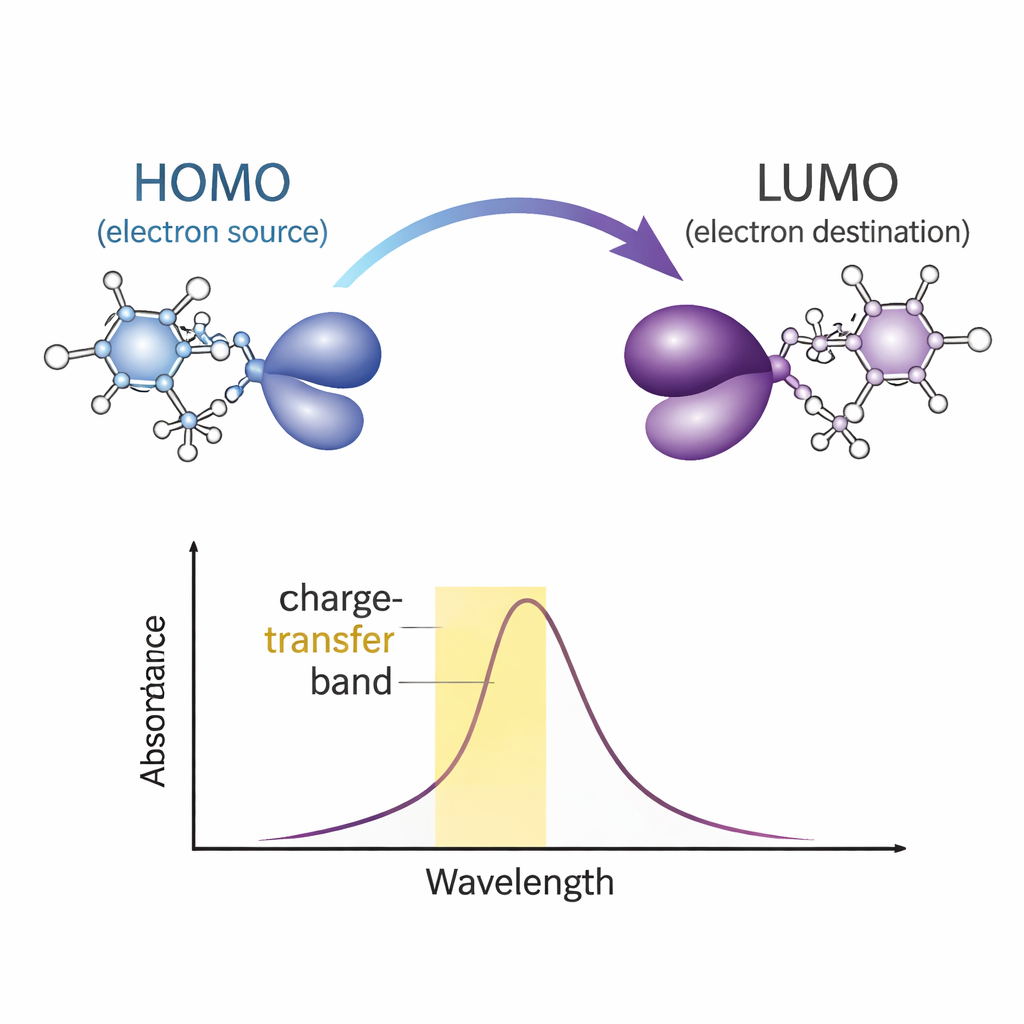
绘制力场与隐藏相互作用
除了简单的成键图像外,作者使用详尽的电子密度分析来观察电荷的实际聚集位置以及弱相互作用如何贡献稳定性。静电势图突出显示电子富集或匮乏的区域,指示每个分子上最易反应的部位并阐明氢键为何在该处形成。自然键轨道(NBO)计算量化了从供体到受体的电子密度流动,确认菲咯啉向对硝基苯酚提供电荷,而对硝基苯酚接受电荷。额外工具,例如简并密度梯度(reduced‑density‑gradient)图与原子-分子拓扑分析,可视化细微的非共价吸引与排斥——范德瓦尔斯接触、氢键与π–π相互作用——这些作用有助于将配合物固定在一起。
从分子细节到光学功能
这一复杂图景的一个特别有前景的结果是对强非线性光学行为的预测:计算表明该配合物对强光场的响应大约比光学中常用的标准参照材料强20倍。通俗地说,这对小型氢键结合的分子能够以对光开关、信号处理和先进光子电路有价值的方式弯曲与混合光。通过明确展示氢键与电荷转移如何重塑结构、电荷分布与光吸收,该研究为设计具有可调电子与光学特性的类似有机配合物提供了配方——这些微小分子模块可能成为未来基于光技术的基础元件。
引用: Hadigheh Rezvan, V., Barani Pour, S., Dabbagh Hosseini Pour, M. et al. DFT study of frontier orbitals and NLO properties of a phenanthroline and nitrophenol complex. Sci Rep 16, 7754 (2026). https://doi.org/10.1038/s41598-026-38340-x
关键词: 电荷转移配合物, 氢键, 非线性光学, 前线轨道, 紫外-可见光谱