Clear Sky Science · zh
在高维空间中使用基于堆的进化框架优化癌症微阵列数据的特征选择
为什么选择合适的基因很重要
基于现代基因技术的癌症检测可以同时测量数以万计的基因,但医生通常只有几十个病人的数据。在这个巨大的“基因丛林”中,真正能将一种癌症类型与另一种区分开来,或将肿瘤与健康组织区分开的,实际上只有少量信号。本文提出了一种新的智能搜索方法,用于自动挑选这些关键基因,旨在使计算辅助的癌症诊断更准确、更快速且更易解释。
信号太多,数据太少
微阵列实验及类似技术使研究人员能够在每个样本中测量数千个基因的表达水平。但样本数量通常很少,有时不足一百。许多基因读数包含噪声、冗余或与所研究疾病无关。保留全部特征会淹没学习算法,降低计算速度,并产生依赖随机特性而非真实生物学的误导性模型。将这些特征裁剪为有用子集的过程称为“特征选择”,这是从高维医学数据中获得可靠预测的关键。
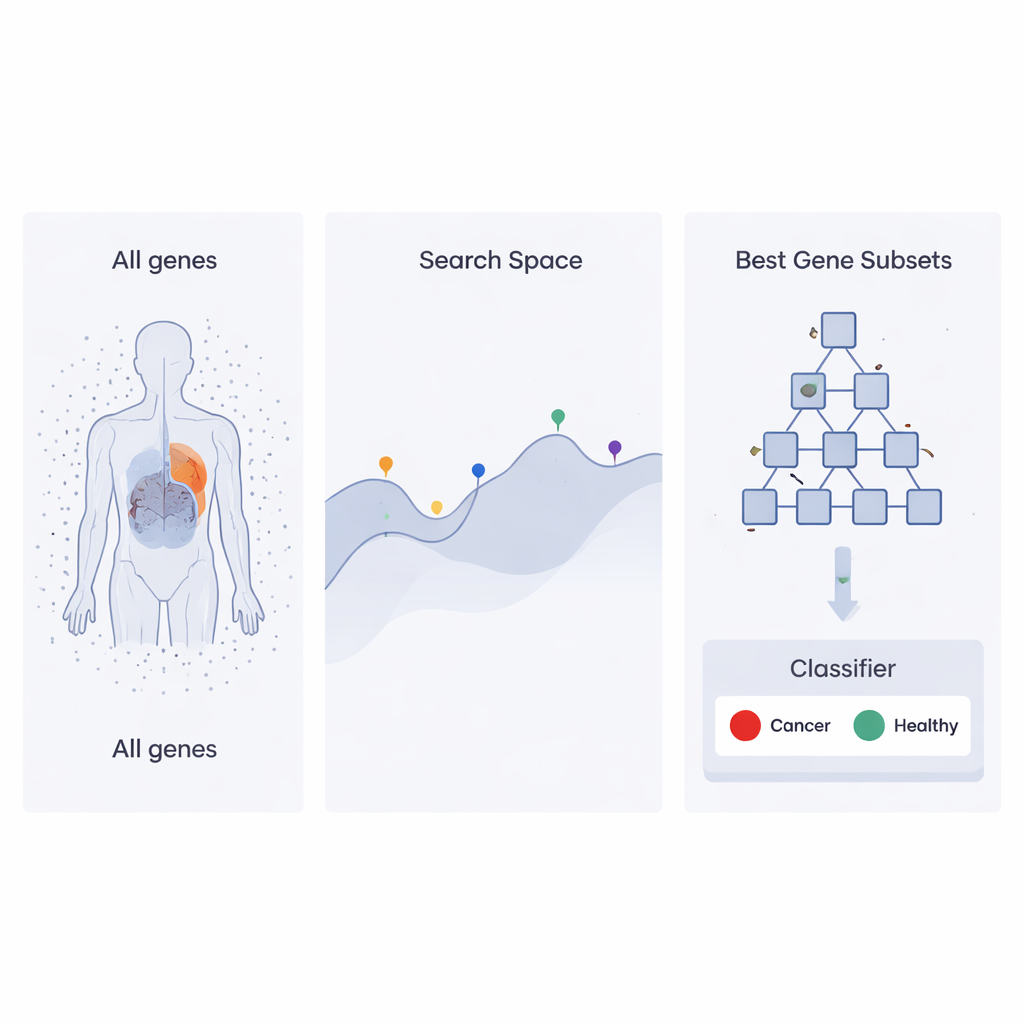
受公司阶梯启发的搜索策略
作者在一种称为基于堆的优化器(Heap‑Based Optimizer,HBO)的近期优化方法基础上进行了扩展,该方法借鉴了企业中员工组织的想法。想象每一种可能的基因集合都是一名“员工”,其工作绩效由该集合在分类器中区分癌症样本与健康样本的效果来评判。这些“员工”使用称为堆的数据结构按层次排列,表现优异的基因集合位于顶部,而较弱的位于下层。经过多轮迭代,低层员工通过复制并略微修改其上级和同事的选择来调整自己,逐步推动整个组织走向更好的解决方案。
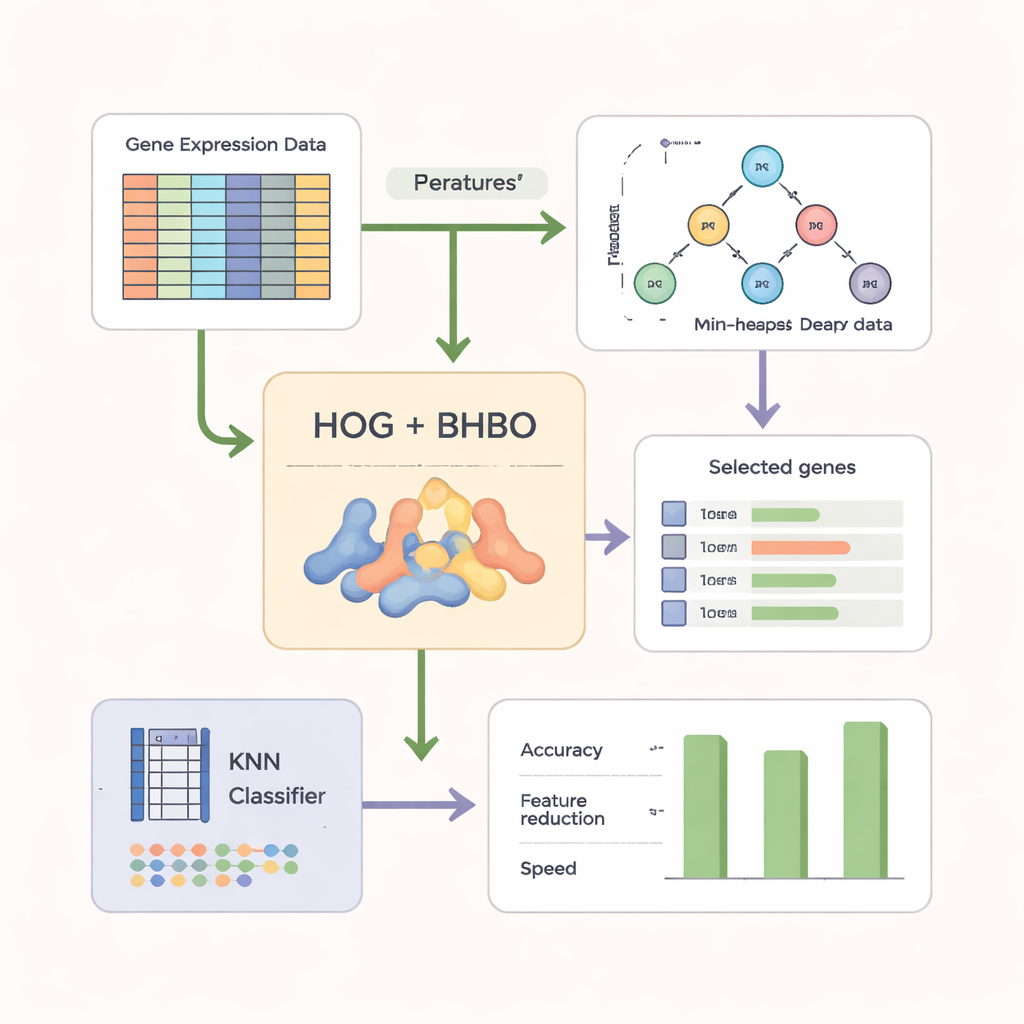
将原始基因数据转化为更清晰的模式
为了提升搜索效果,作者不单靠原始基因读数。他们首先将微阵列数据重新构造成类似图像的形式,并应用在计算机视觉中广泛使用的方向梯度直方图(Histogram of Oriented Gradients,HOG)技术。HOG 捕捉基因表达水平随基因位置变化的模式,强调局部模式而非孤立的测量值。基于这些模式的特征随后与原始基因信息结合。一个简单的分类器——k 最近邻(KNN)——作为“裁判”,根据候选基因子集对新样本标注的准确性进行评分,同时还对更小、更紧凑的基因集合进行奖励。
在多种癌症数据集上的测试
研究人员在九个公开的癌症微阵列数据集上评估了他们的二元化基于堆的优化器(BHBO),包括脑肿瘤、白血病、前列腺癌以及包含多个亚型的混合肿瘤集合。每个数据集测量的基因数量从数千到超过一万五千不等,但病人样本较少。对于每个数据集,BHBO 被重复运行并与七种知名搜索方法(例如遗传算法和粒子群优化)进行比较。团队不仅测量了分类准确率,还评估了保留的基因数量、搜索收敛速度以及在模拟噪声、批次效应和标签错误干扰下结果的稳定性。
新方法取得的成果
在这九个数据集中,基于堆的方法平均达到约 95% 的分类准确率,同时将基因数量减少了超过 85%。在若干数据集上,它明显优于竞争方法,并显示出更快的收敛性——即在更少的搜索步骤内找到良好的基因集合。即便作者故意破坏数据——加入噪声或翻转部分样本标签——该方法的性能仅略有下降且仍优于替代方法。统计检验证实,这些改进不太可能由偶然造成。
对未来癌症诊断的意义
在实践层面,这项工作表明,经过精心设计的搜索策略可以筛选庞大的基因数据集,发现仍能很好地分类癌症的小型高信息量基因面板。对于临床医生和研究人员而言,这类紧凑的基因集合更易于生物学验证、后续检测成本更低,并更适合集成到决策支持工具中。尽管该方法并不直接发现新药物或新通路,但它将聚光灯聚焦到有价值的基因标记上,帮助后续研究关注隐藏在高维癌症数据中最具信息性的信号。
引用: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
关键词: 癌症微阵列, 特征选择, 元启发式优化, 基因生物标志物, 医学数据挖掘