Clear Sky Science · zh
一种针对白对虾基因组结构的适应性框架,用于基于ROH的近交估计
为什么对虾家谱与餐桌息息相关
现代虾场供应了全球大量海鲜,但重复使用相同的家系育种会在不知不觉中削弱种群健康。当近亲交配发生时,隐性的有害基因可能配对出现,导致生长、存活率和抗病力下降。本研究提出了一个看似简单但对全球水产养殖意义深远的问题:如何足够准确地测量养殖对虾的近交程度,以保持种群健康和生产力?
近交的隐性基因足迹
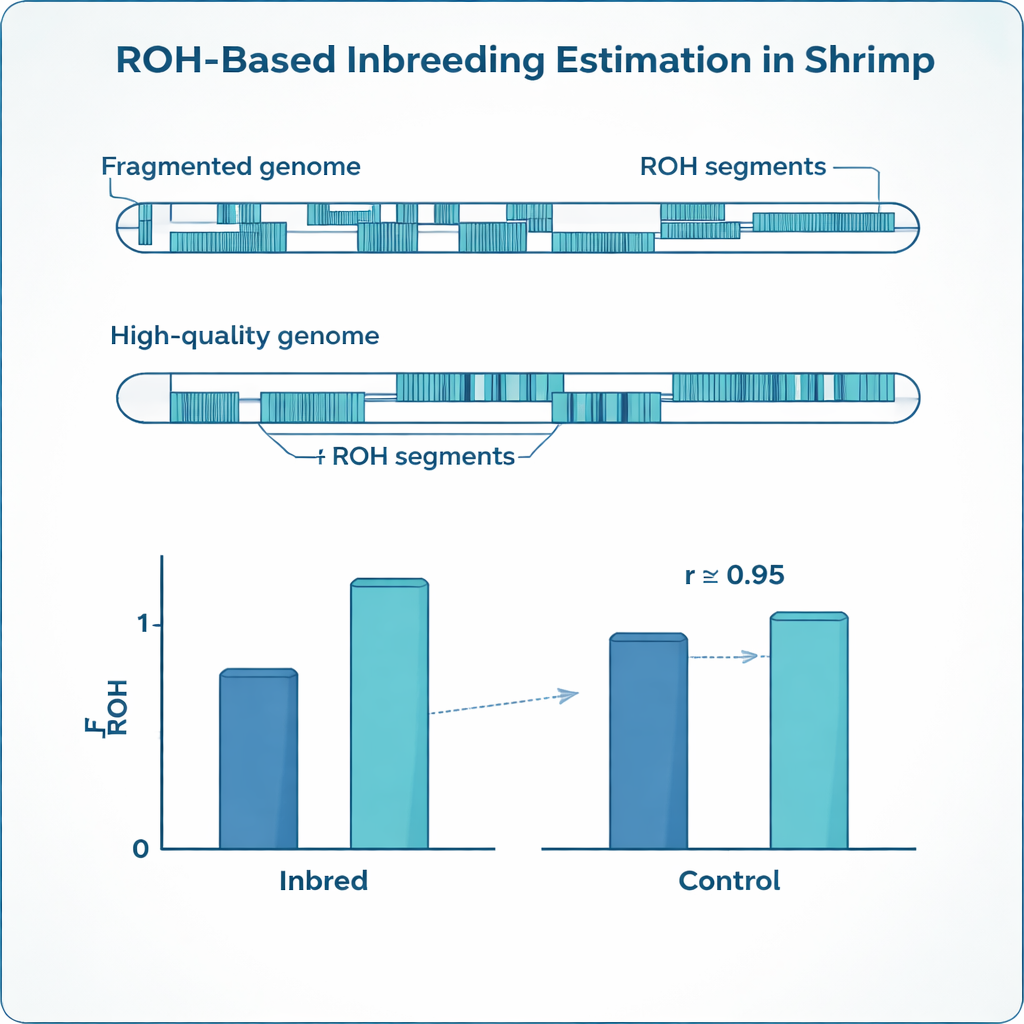
近交会在DNA中留下明显的痕迹。与携带两种略有不同的基因拷贝相比,近交动物往往在较长的片段上两条拷贝相同。遗传学家将这些片段称为“全合子片段”(runs of homozygosity,ROH)。通过累加个体基因组中属于这些片段的比例,研究者可以比依赖常有遗漏或错误的纸质谱系更精确地估计近交程度。这种基于ROH的测量,称为FROH,已成为牛、猪等畜牧业的标准方法,但对虾的基因组具有特殊挑战,使得现成的方法往往不够可靠。
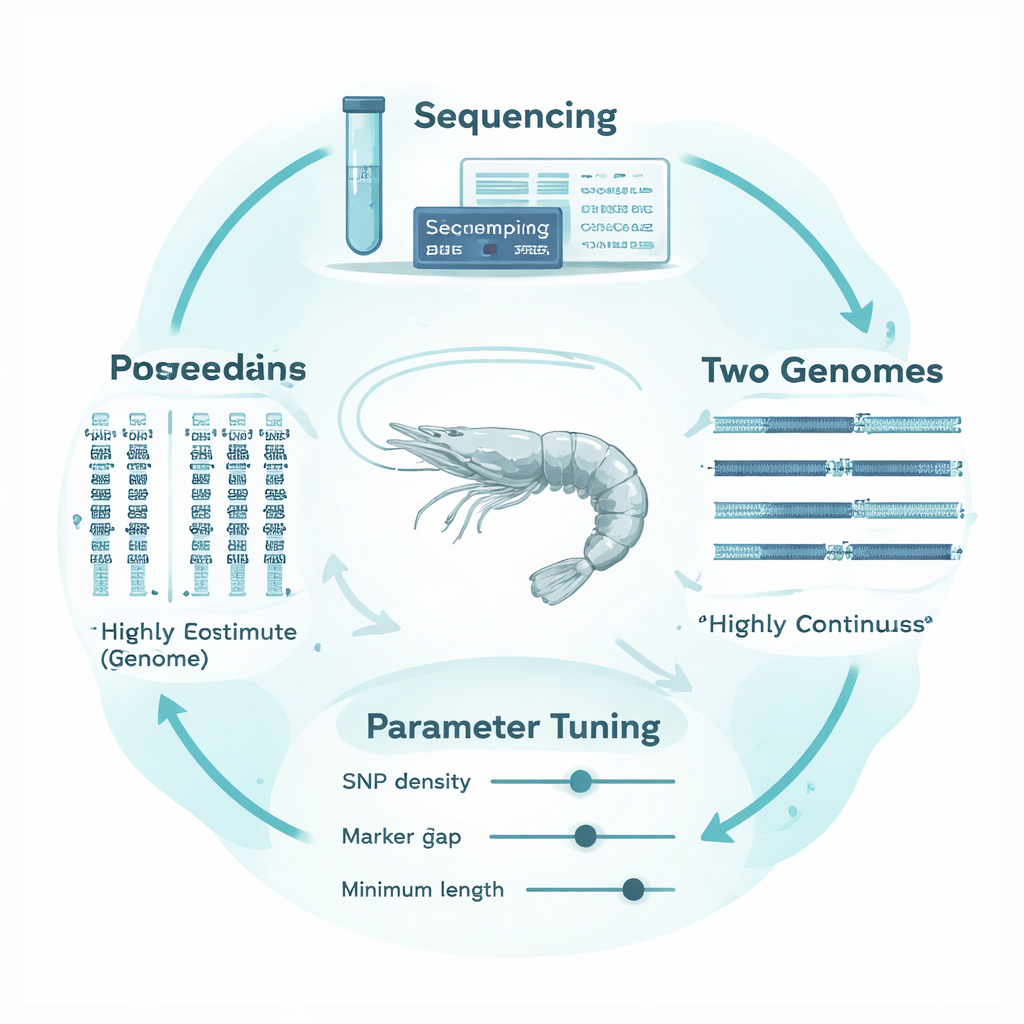
为什么对虾基因组尤其棘手
白对虾(Penaeus vannamei)是全球养殖最广泛的对虾,其基因组高度分裂且结构复杂。与连续的染色体序列不同,现有的许多基因组组装被拆成数千个小片段,间隔有缺口和重复区域。遗传标记在这块拼凑的地图上分布不均,而且该物种具有很高的遗传多样性。因此,原本为结构整齐、组装良好的哺乳动物基因组调优的方法和软件设置,可能会把技术性缺口误判为ROH的真实断裂,或将短的背景相似性误认为近交的信号。结果是极有可能错误估计对虾的实际近交程度。
构建一个考虑基因组结构的测量框架
为了解决这一问题,作者设计了一个“基因组结构适应性”框架,根据对虾DNA的特点调整ROH分析流程。他们在受控交配下建立了13个高度近交的虾系,并对其中5个家系及其亲本进行了深度测序。关键在于,他们将相同的测序数据分别比对到两个非常不同的参考基因组:一个较早的、分裂严重的组装和一个更新的、高度连续的版本。使用常用分析工具PLINK,系统地测试了8个关键参数如何影响ROH的判定,重点关注与基因组结构特别相关的三个参数:标记的最小密度、允许在片段内出现的标记间最大间距以及计入的最短片段长度。他们建立了经验性的、不重叠的基因组窗口来跟踪局部标记间距和缺失数据,并使用这些窗口的“基因组覆盖度”以及FROH和ROH长度的稳定性,作为选择合理阈值的客观依据。
不同的参数设置,趋同的近交结论
优化后的参数对于两个参考基因组差异很大。相比高质量的连续组装,分裂的基因组需要更高密度的标记、更短的允许间距和更短的最小片段长度,以避免将真实的ROH切分成许多小片段。然而,在分别微调这些参数后,两种参考给出的近交估计达成了趋同:近交虾的平均FROH在两种情况下均约为0.24,与计划交配的预期值高度一致且彼此强烈一致。与此同时,更连续的基因组显示的ROH数量较少但每段更长,而分裂的地图则把许多长片段分割成短的区间。研究还揭示了同窝全同胞之间存在显著差异:即使在同一家系内,个体间的近交程度也差异很大,这是简单谱系记录无法捕捉到的。
更精确的工具,助力更健康的虾种群
对非专业读者来说,关键结论很直接:用DNA测量养殖对虾的近交可以非常精确,但前提是方法必须尊重基础基因组的结构。通过提供一套实用的流程来针对分裂的甲壳动物基因组调整ROH分析,这项工作使育种者能够逐个体监测近交,而不必依赖不完备的家谱记录。反过来,这有助于设计既能保持遗传多样性又能提升生长和抗逆性的配种方案,支持更可持续的对虾养殖,并为面临类似基因组问题的其他水产物种提供范例。
引用: Zou, X., Zhou, H., Liu, M. et al. A genome-structure adaptive framework for ROH-based inbreeding estimation in Penaeus vannamei. Sci Rep 16, 6769 (2026). https://doi.org/10.1038/s41598-026-37622-8
关键词: 对虾育种, 近交, 基因组选择, 全合子片段(ROH), 水产养殖遗传学