Clear Sky Science · zh
自适应模糊簇引导的简单、快速且高效的特征选择方法,适用于高维且高度不平衡的二分类生物信息学微阵列数据
这对基因研究为何重要
现代基因表达检测可以在单一患者样本中测量数以万计的基因。这种海量数据有望带来更早的癌症诊断和更明智的治疗选择,但也带来问题:大多数基因是噪声、冗余,或主要与常见病例相关,而非那些罕见且危险的病例。本文提出了一种新的方法,在庞大的基因表达数据集中进行筛选,使计算机能够仅凭极少且精心挑选的基因,可靠地识别出处于小而难以检测的少数患者群体。
太多且过于相似的基因带来的挑战
微阵列实验通常对仅有数百名患者的样本追踪数千个基因的活动水平。通常,一个类别(例如常见的癌症亚型)远多于另一个,导致数据高度不平衡。在这种情况下,许多基因表现出非常相似的行为,多数类与少数类的模式可能重叠。标准的学习方法往往偏向多数类,并被冗余基因所迷惑,从而导致过拟合并难以检测罕见亚型。传统的降维方法要么通过构建新的混合特征而失去可解释性,要么在选择基因时没有仔细考量这些基因对分类器识别少数类的实际帮助。
更聪明的基因选择新路线图
作者提出了AFCG‑SFE,一种专为高维、不平衡的基因表达数据设计的自适应特征选择模型。该方法从一个简单的“单智能体”搜索开始,该搜索将基因开/关并测试其对分类的支持度,但通过若干数据驱动步骤加以增强。首先,根据基因的行为相似性将其分组,并允许基因属于多个组,以反映基因可能参与多条生物通路的生物学现实。在每个簇内,根据基因对疾病标签的信息量对其进行排序,仅保留少数关键代表,从而在主搜索开始前就显著削减冗余。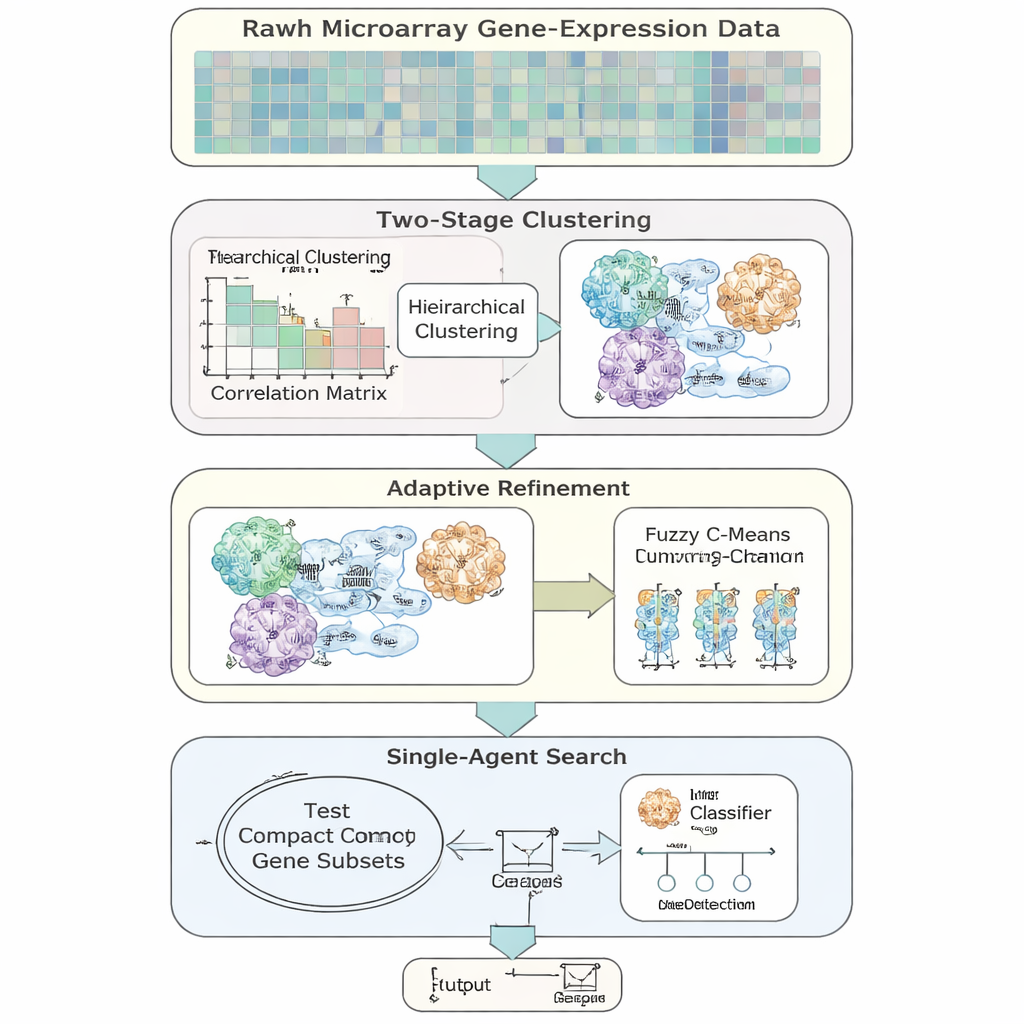
使算法关注稀少患者
AFCG‑SFE并非仅关注简单的准确率,而是使用一种强调适用于偏斜数据的指标的适应度评分,包括在正确识别少数类与多数类之间的平衡以及在所有决策阈值下的表现。该适应度函数还对选择过多基因或来自同一簇的过多基因施加惩罚,并对与疾病标签具有强依赖性的基因给予奖励。重要的是,这些惩罚和奖励的强度由数据集的属性(例如每名患者的基因数和类别间的重叠程度)自动设定,而不是通过手动调参。这使得该方法更稳健,更容易在不同研究之间迁移。
适应问题难度
一个关键思想是算法不应总是追求尽可能最小的基因集合。当两个类别难以区分或高度重叠时,该方法会自动提高必须保留基因的下限,确保不会丢弃罕见但重要的信号。随着搜索的进行,AFCG‑SFE逐步收紧每个簇中允许存活基因的上限,同时仍然尊重这一最小值。其结果是一个紧凑且多样化的基因面板,能够捕捉数据的结构而不被单一冗余模式主导。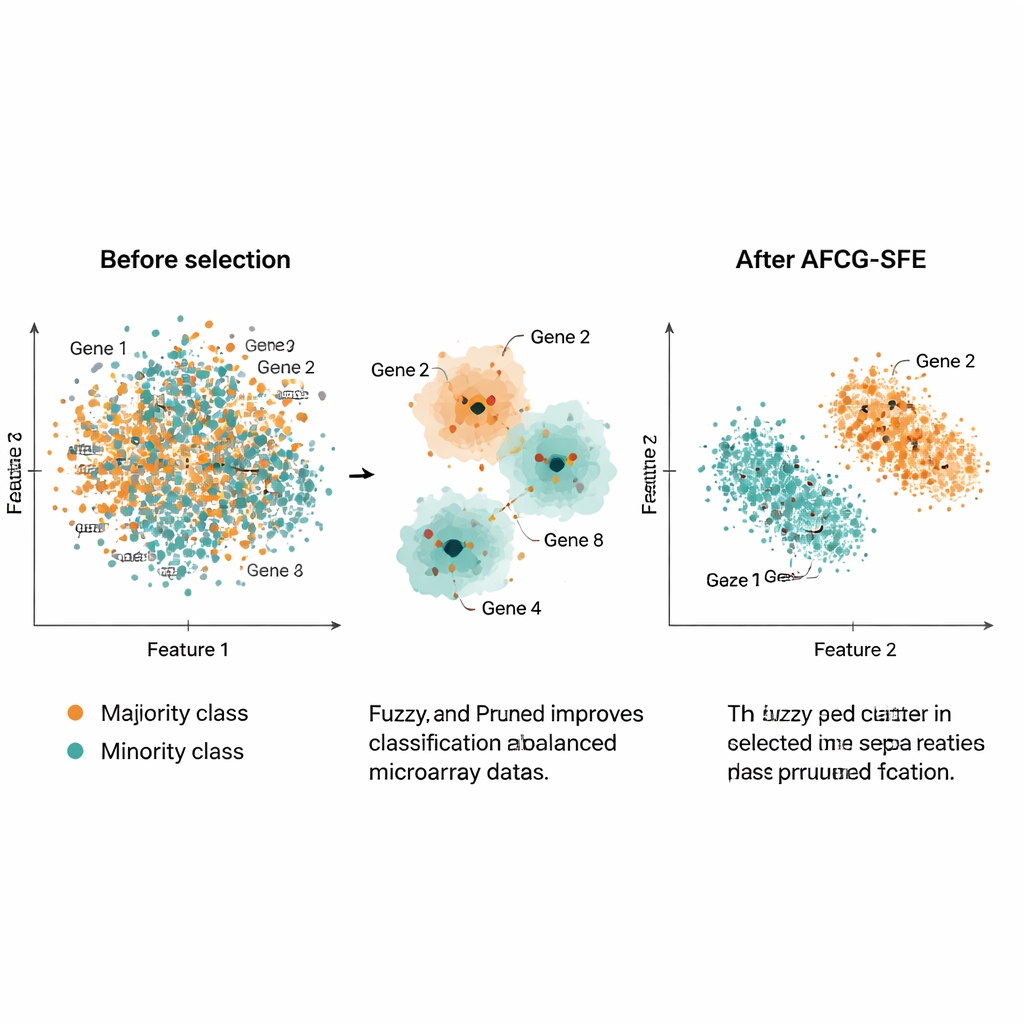
实验结果显示了什么
作者在20个公开的癌症微阵列数据集上测试了AFCG‑SFE,每个数据集均具有数千个基因但仅约100–200个样本且存在强烈的类别不平衡。他们将该方法与若干进化搜索基线、简单滤波器和将特征选择嵌入分类器的嵌入式方法进行了比较。在包括F值、平衡准确率、ROC曲线下面积以及过拟合度量在内的一系列指标上,AFCG‑SFE在所有数据集上均为最佳或并列最佳。它通常选择少于25个基因(常见为6–8个),在仍能改善或维持分类性能的同时,去除了超过99%的原始特征。它还降低了一个反映类别在特征空间重叠程度的复杂性指数,表明选择后类间分离更为清晰。
非专业读者的结论
从实用角度看,这项工作提供了一种将庞大且嘈杂的基因表达谱缩减为极小且有信息量的基因集合的方法,同时仍能使计算机准确识别罕见患者亚群。通过智能地分组相似基因、奖励真正与疾病相关的基因,并明确防止对多数类的偏倚,AFCG‑SFE在提升预测能力的同时显著简化了基因面板。这种组合有助于研究人员锁定潜在生物标志物、设计更具可解释性的诊断测试,并最终改善精准医学工具在真实且不完美生物数据上的表现。
引用: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
关键词: 基因表达, 特征选择, 不平衡数据, 微阵列, 癌症亚型