Clear Sky Science · zh
rhinotypeR 可从 VP4/2 序列实现可重复的鼻病毒基因分型
为什么微小的感冒病毒依然重要
我们大多数人把普通感冒视为一种恼人的小毛病,而非严重威胁。然而,引起许多感冒的病毒——人类鼻病毒——也与严重肺部感染、哮喘发作以及慢性肺病的恶化有关。为了追踪这些病毒如何演化与传播,科学家需要将它们划分为精确的遗传“类型”,类似于为商品贴上条码。本文介绍了 rhinotypeR,这是一款免费、开源的软件包,能使这种遗传标注更准确、一致且易于重复,帮助公共卫生团队更清晰地监控这一常被忽视的呼吸道病毒家族。
普通感冒中的隐性多样性
人类鼻病毒极为常见,在急性呼吸道疾病患者的样本中可占到多达 60%。它们并非单一病毒,而是分为 A、B、C 三个主要群组,且至少有 169 个被认可的遗传类型。不同类型的表现各不相同:有些类型更常与儿童的严重感染和哮喘加剧相关,而另一些在重症中则较少见。由于这些类型各自独立演化并携带不同的表面特征,科学家若要跟踪疫情如何在学校、家庭和社区间传播,就需要可靠的方法将它们区分开来。
从零散工具到统一流程
迄今为止,从基因序列判定鼻病毒类型一直是个拼凑式的工作。研究人员通常聚焦于病毒基因组中一段称为 VP4/2 的短序列,将其与已知参考株比对,测量序列间的差异,再使用阈值来决定每个样本属于哪个类型。但这些步骤往往由多种软件、人工编辑和个人判断混合完成,导致不同研究之间即便使用相似数据也难以比较或重复。rhinotypeR 的诞生正是为将这一多步骤、容易出错的过程转化为任何人都能运行并共享的单一脚本化工作流。
新软件的实际功能
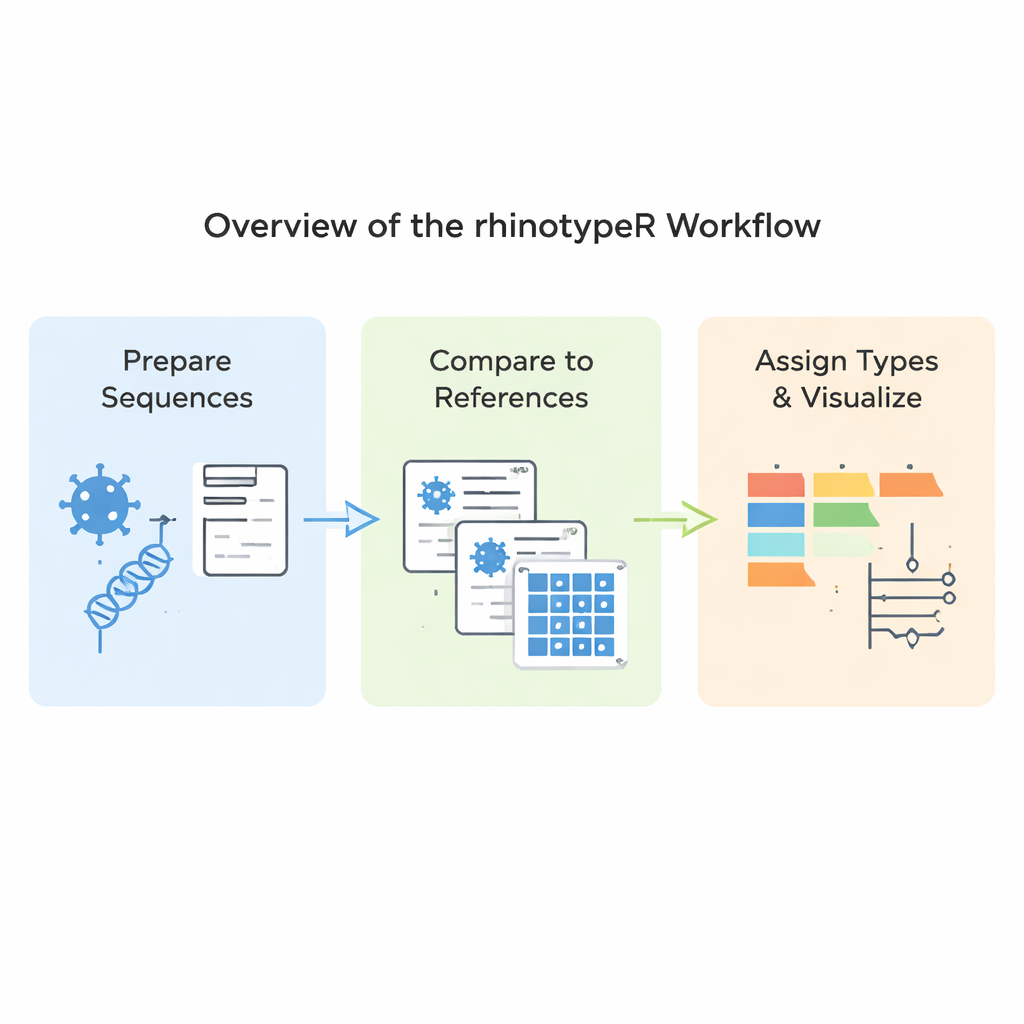
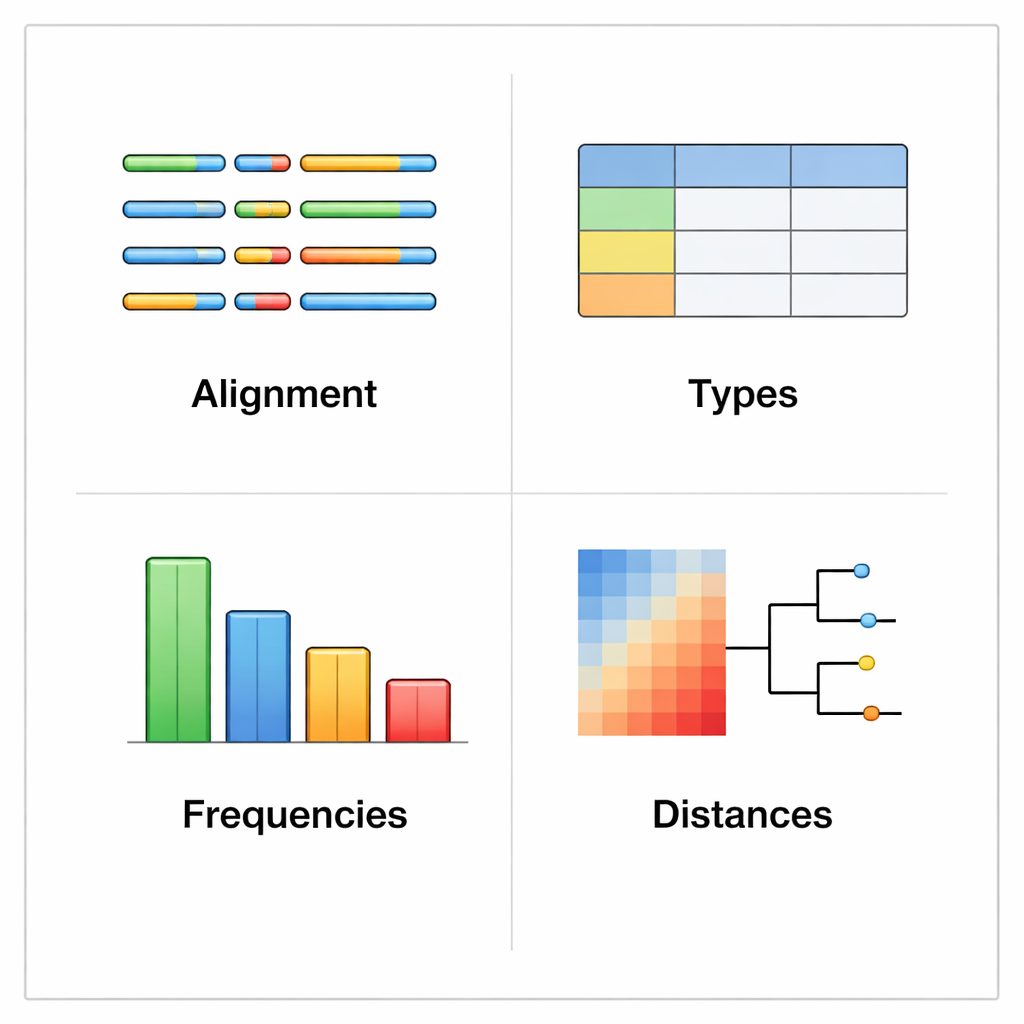
rhinotypeR 在广泛使用的 R 与 Bioconductor 分析环境中运行。它接受一组鼻病毒 VP4/2 序列,并通过三个主要阶段处理:准备并比对序列、计算每条序列与经人工整理的参考类型之间的遗传距离,以及将每个样本分配给最接近的已知类型,或在差异过大时标记为“未分配”。该工具还可生成可视输出,包括以颜色标示的遗传差异图、简明的谱系树,以及显示每种类型在数据集中频率的图表。用户可选择用外部程序进行比对,或让 rhinotypeR 在 R 内部完成整个流程以获得最大可重复性。
对工具的检验
为了验证 rhinotypeR 的结果可信,作者将其距离测量结果与两个已建立程序(ape 与 MEGA X)进行比较,使用相同的输入文件和模型。结果几乎完全一致;任何微小差异都源于计算四舍五入等常见数值误差,而非方法本身不同。团队随后在来自多项既有研究的超过 2,300 条鼻病毒序列上运行 rhinotypeR,涵盖了已知类型的 90% 以上。在大约五分之四的情况下,该工具与先前的类型标注完全一致。大多数分歧发生在用于区分类型的预先设定阈值附近,正是边界判定最容易出现争议的地方。重要的是,无法被自信分配到已知类型的样本并未表现出质量差或病毒量低的迹象,这表明它们可能反映了真实的病毒多样性。
这对公共卫生的重要性
对非专业读者来说,关键在于 rhinotypeR 并未重新发明科学家分类感冒病毒的方法;相反,它使该过程更清晰、透明且易于重复。通过将比对、距离计算和类型分配打包到一个开源软件包中,并提供清晰的可视化汇总,它帮助研究人员和监测项目以一致的方式处理数千个样本。这种一致性提升了我们比较不同时间和地点研究的能力,使我们更早识别异常或新兴的病毒谱系,并将遗传模式与现实世界的疾病趋势联系起来。从长期看,像 rhinotypeR 这样的工具能强化对看似普通但在许多人中能引发严重疾病的感冒的常规监测。
引用: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
关键词: 鼻病毒分型, 分子监测, VP4/2 测序, 生物信息学工具, 呼吸道病毒