Clear Sky Science · zh
通过实时监测酶促反应(结合核磁共振与计算方法)研究酶的功能热点残基
这对未来抗病毒药物为何重要
法匹拉韦是一种已用于流感并在 COVID-19 中接受测试的口服药,但它以我们摄入的形式并不直接对抗病毒。它必须首先由机体细胞转化为具有阻断病毒活性的分子。本研究几乎逐原子地解析了人体中一种酶如何完成这一关键的活化步骤,以及酶中哪些微小部位充当控制药物开启速度与效率的“热点”残基。理解这些细节有助于指导下一代抗病毒药物的设计,使其在效力与临床表现上更为可预测。
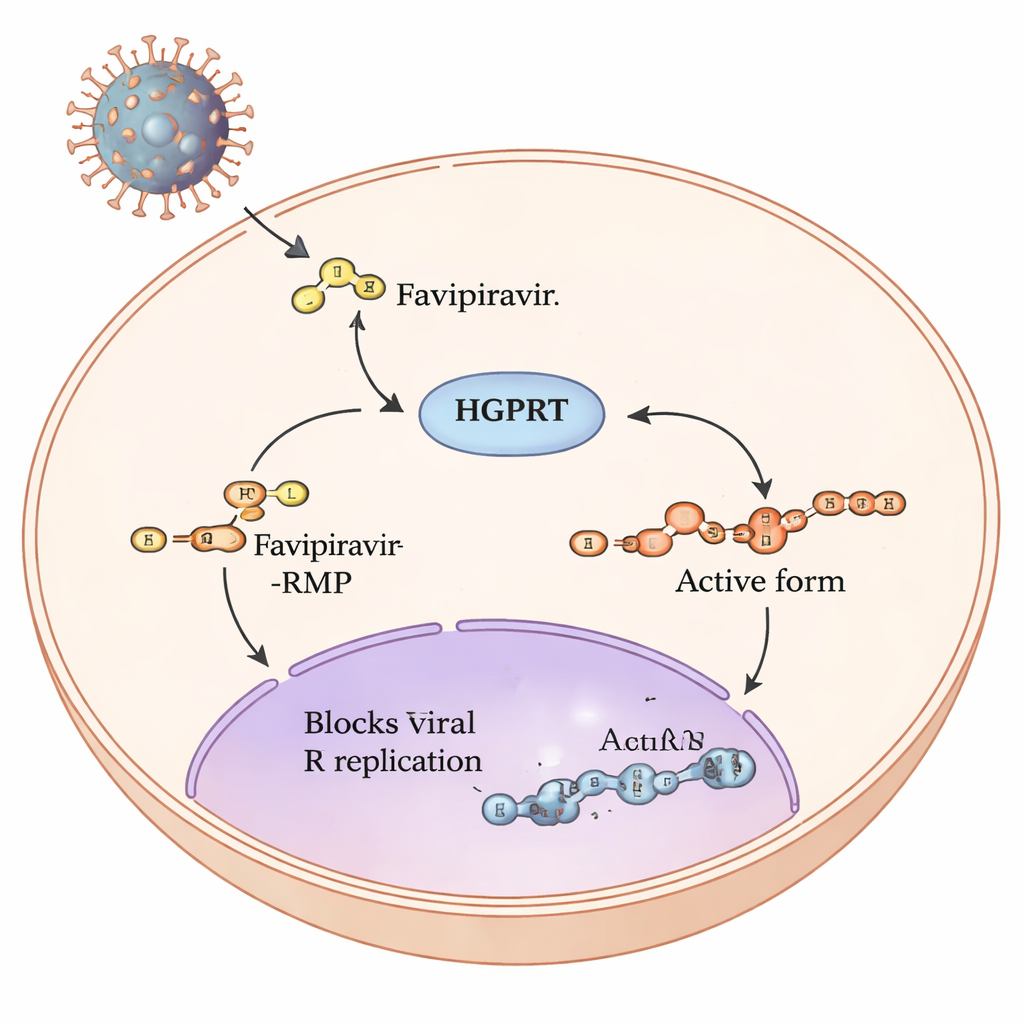
前药在细胞内的演变过程
法匹拉韦属于所谓的前药:进入人体细胞后,一系列化学步骤将其改造成能够干扰 RNA 病毒(如流感病毒与 SARS‑CoV‑2)复制机器的形式。该途径的第一个也是最慢的一步由人类酶次黄嘌呤‑鸟嘌呤磷酸核糖转移酶(HGPRT)完成。HGPRT 向法匹拉韦添加一个小的糖‑磷酸基团,生成法匹拉韦‑RMP。只有在完成这一步之后,其他酶才能进一步构建出直接干扰病毒 RNA 聚合酶的三磷酸活性形式。由于 HGPRT 驱动的这一步像瓶颈一样限制了活性药物的产生量,作者们致力于确定 HGPRT 中哪些部位对处理法匹拉韦最为关键。
用核磁实时观察化学反应
法匹拉韦独特地包含一个氟原子,在磁场中类似微小无线电发射器。团队利用这一点,采用 19F 核磁共振(NMR)波谱学实时监测反应过程中试管中法匹拉韦与法匹拉韦‑RMP 的含量。由于只有药物分子含氟,NMR 信号清晰、易于追踪。通过在 12 小时内重复记录光谱,研究者们能够跟踪起始药物的消失与产物的生成,然后提取常规动力学参数,例如反应速率和酶对药物的表观结合紧密度。
调控酶中的关键位点
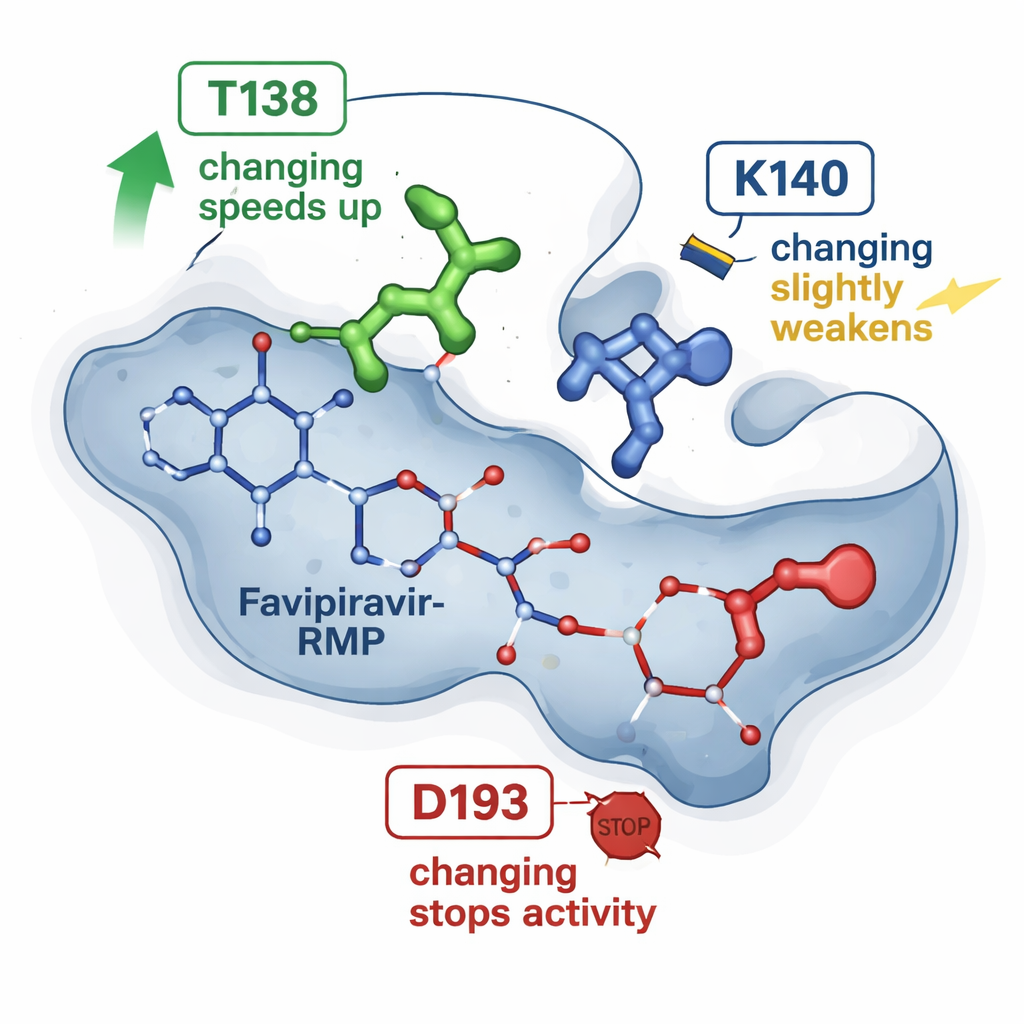
先前 HGPRT 与法匹拉韦‑RMP 结合的 X 射线结构提示了少数将药物包裹在口袋中的氨基酸。新研究通过在蛋白中进行精确的单个字母替换来检验其中三个位点,并将每种突变体酶与天然酶进行比较。一处替换(记为 T138A)出人意料地使酶将法匹拉韦转化的速度提高了约四到六倍,尽管它去除了一个此前被认为有助于固定药物的化学基团。第二处替换 K140M 使反应稍微变慢并略微削弱了表观结合。第三处替换 D193N 则完全丧失了酶合成法匹拉韦‑RMP 的能力,尽管改造后的蛋白仍能被表达并与产物结合。这些结果表明,并非所有接触点都是等同的:有些充当微妙的速率调节器,而另一些则是必需的开关。
在计算机上模拟运动部件
为了超越静态结构,研究人员转向分子模拟。从已知的 HGPRT 与法匹拉韦‑RMP 的三维结构出发,他们使用成熟的计算工具估算每种突变体中药物的结合强度,并运行大量短时的分子动力学模拟。这些模拟追踪原子在数十纳秒尺度上的振动与相互作用。计算结果与 NMR 得到的趋势一致:T138A 变体倾向于更有利地保持法匹拉韦‑RMP,但也出现药物沿“逃逸”通道移动的片段,而另一个残基(K140)短暂锚定磷酸基后释放该组分。相比之下,D193N 变体仍能抓住产物,但可能在需要镁离子的早期催化步骤上失败,这解释了尽管结合稳定却失去活性的原因。
更聪明抗病毒设计的路线图
通过将实时 NMR 测量与细致的计算模型结合,这项研究绘制了 HGPRT 中控制法匹拉韦活化效率的功能热点图。对非专业读者而言,结论是:我们自身的酶会显著影响细胞内活性抗病毒药物的积累量,改变药物的形状或酶的口袋都可能显著改变这一结果。作者的混合策略为探究其他药物如何与其目标蛋白相互作用提供了通用蓝图,或可加速开发与机体活化机制更匹配的新型抗病毒化合物。
引用: Sugiki, T., Yoshida, T., Tsukamoto, M. et al. Investigation of the functional hot-spot residues of an enzyme by real-time monitoring of the enzymatic reaction using NMR and computational approaches. Sci Rep 16, 5896 (2026). https://doi.org/10.1038/s41598-026-35354-3
关键词: 法匹拉韦, 抗病毒活化, HGPRT 酶, 核磁共振波谱学, 药物设计