Clear Sky Science · zh
用于极高维基因组数据的随机 LASSO
在基因组干草堆中寻找针
现代生物学可以同时测量数以万计的基因,但患者研究通常只包含几百人。在这种不平衡中,隐藏着少量真正对预测疾病风险或生存期有意义的基因。本文提出了“随机 LASSO”(Stochastic LASSO),这是一种统计方法,旨在从大量噪声的基因组数据中可靠地发现这些关键基因,即使基因数量远多于患者样本数也能有效工作。
为什么挑选合适的基因如此困难
研究人员常用的工具之一是 LASSO,它将不重要的基因效应压缩为接近零,同时保留最有信息量的基因。然而,在基因数量远超样本数量(如癌症基因组学中常见)的情况下,经典 LASSO 表现不佳。标准 LASSO 最多只能选择与样本数相当数量的基因,而且往往会忽略行为相似的基因。早期通过增加额外惩罚项的改进能处理部分相关性,但可能会模糊生物学含义,迫使相关基因表现得好像它们都以相同方向影响结局。
构建更“干净”的随机子样本
一种有前景的解决办法是对许多较小的、随机抽取的基因子集重复拟合 LASSO,然后将结果合并。然而,这些“自助法”(bootstrap)方法仍面临三类问题:相关基因可能相互抵消,许多基因很少或从未被抽到,以及纯随机性使最终选择不稳定。随机 LASSO 通过一种称为基于相关性的自助采样的新抽样方案正面解决这些问题。它不是完全随机地选基因,而是有意偏向那些与已有已选基因相关性较低的基因,从而产生更小且更独立的基因集合。它还确保每个基因在所有自助运行中被使用的次数相同,避免某些基因被不公平地忽视。 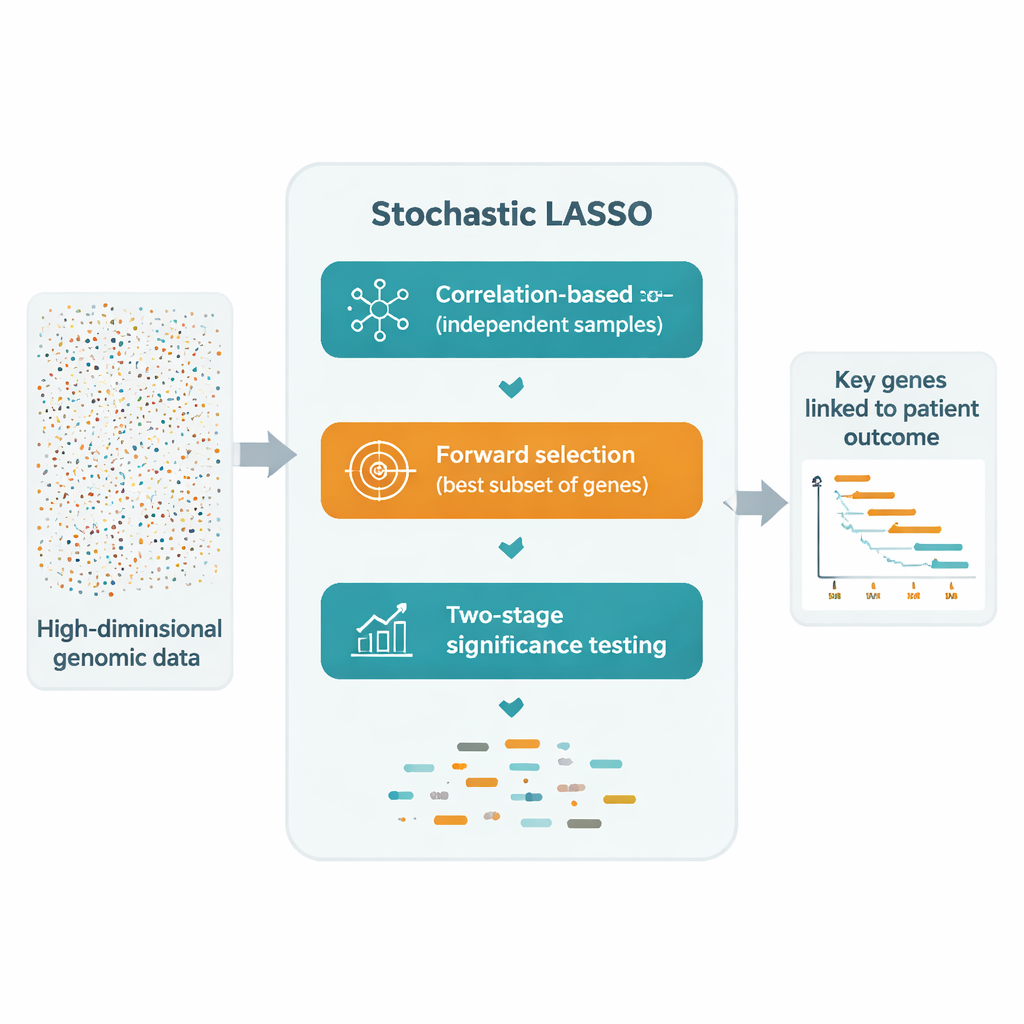
从局部线索到全局基因集
在构建这些更“干净”的子集后,随机 LASSO 记录每个基因在所有自助拟合中的系数大小。这一平均绝对效应成为反映基因一致重要性的“局部得分”。方法并不穷尽地测试每一种可能的组合,而是按局部得分的顺序逐步添加基因以构建候选模型,并在独立的验证数据上评估每个候选模型的预测性能。通过这种方式,它最终选择出一组紧凑的基因集合,这些基因的组合信号最好地解释数据,同时所需的尝试次数远少于传统的逐步方法。
检验哪些基因确实重要
为了将“经常被选中”转化为“统计上令人信服”,作者引入了一个两阶段 t 检验。首先,检查每个基因在自助样本中平均系数是否显著不同于零,将其标记为潜在有意义基因。然后,在这些候选基因中,进一步检验每个基因的效应是否大于所有候选基因的典型效应大小。只有同时通过两项检验的基因才被判定为显著。由于这些检验依赖大量的自助估计,随机 LASSO 能够自信地识别出比患者数量更多的显著基因——这是传统 LASSO 无法做到的。 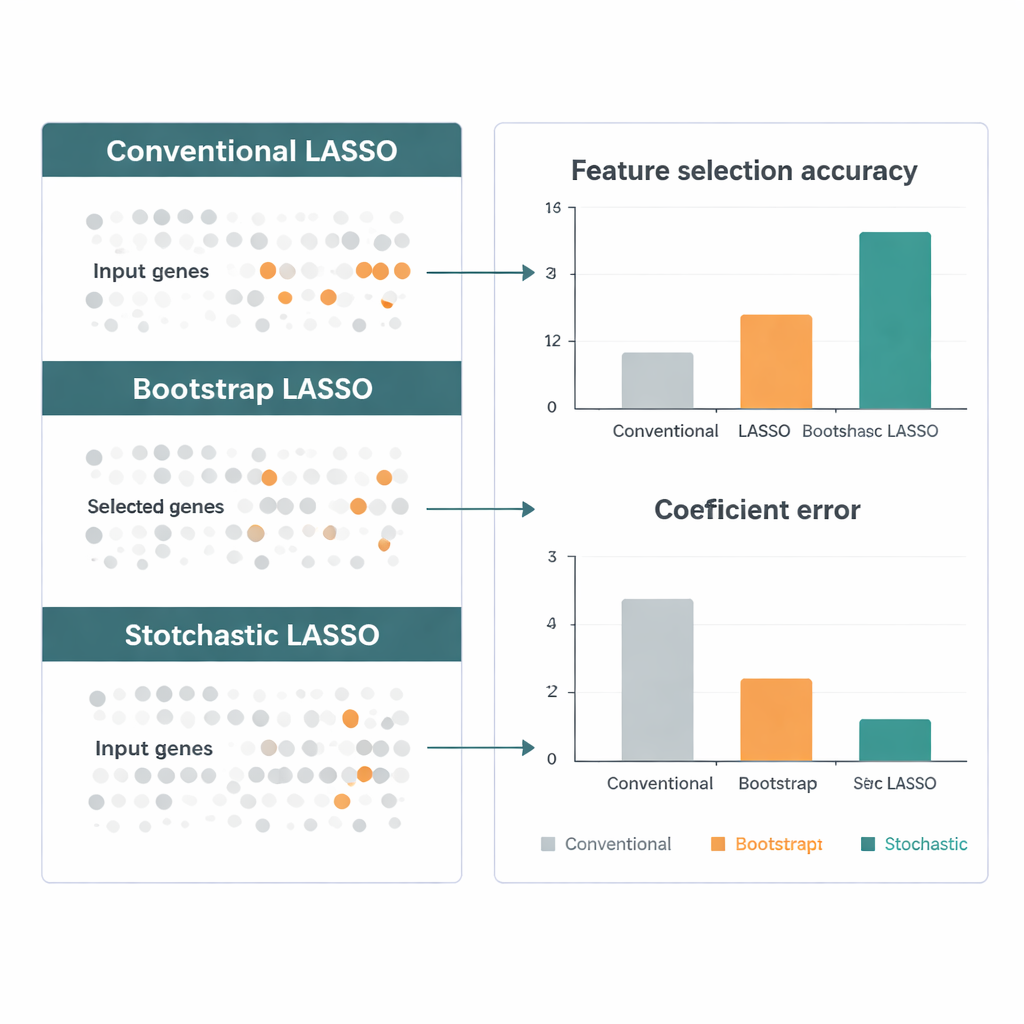
在模拟和癌症数据中证明其价值
作者将随机 LASSO 与若干领先的 LASSO 变体在模拟数据上进行基准比较,模拟数据旨在模拟真实基因组研究的情形:基因数量极多、相关性强且已知“真实”信号。在多种情形下,新方法更频繁地找到正确基因、更准确地估计其效应,并且从一次运行到下一次保持稳定。随后他们使用来自癌症基因组图谱(TCGA)的脑肿瘤基因表达数据进行验证,包括侵袭性胶质母细胞瘤。随机 LASSO 强调了数百个与患者生存相关的基因,并指出了生物通路——例如信号传导和药物代谢途径——这些通路在文献中具有独立支持,表明该方法不仅在统计上更为敏锐,而且在生物学上也合理。
这对患者和研究者意味着什么
对非专业读者而言,关键结论是,随机 LASSO 是一种更智能的基因组大数据筛选工具。它帮助科学家在数据有限且基因高度相互关联的情况下,将真正与疾病相关的基因从统计噪声中分离出来。通过提供更准确且更稳定的基因列表和效应估计,它可以为癌症和其他复杂疾病中的生物标志物、药物靶点和预后特征的搜索提供更清晰的方向。尽管该方法在文中以线性回归为示例,但相同框架可以嵌入生存模型和分类问题,扩大其在生物医学研究中的潜在影响。
引用: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
关键词: 基因组特征选择, 高维数据, LASSO 方法, 癌症基因表达, 生物标志物发现