Clear Sky Science · zh
大流行规模系统发育学中的速率变异与重复序列错误
这对未来疫情为何重要
当一种新病毒在全球传播时,科学家争相读取其遗传密码并重建其家族树。这些树有助于追踪变体如何出现、传播速度以及防控措施是否有效。但在 COVID-19 期间,各实验室在极短时间内测序了数百万份 SARS‑CoV‑2 基因组,数据中隐藏的错误和特性开始扭曲整体图景。本文引入了用于清理和解释如此庞大遗传数据集的新方法,从而更清晰地呈现大流行病毒如何在种群中真正演化与扩散。
在数百万个基因组中理清头绪的挑战
基因组流行病学将病毒基因组转化为可供公共卫生决策使用的实用信息。对于 SARS‑CoV‑2,全球已共享超过两千万份基因组。传统的进化工具最初为更小规模的问题设计,例如在物种之间比较基因,而非处理实时到达的、彼此高度相似的数百万条病毒序列。在这一规模上,有两个问题尤其棘手。首先,病毒基因组中的某些位点比其他位点变异频繁,这会使无关的病毒看起来异常相似。其次,测序与数据处理过程中的重复技术性错误可能会模拟真实突变。这两种效应在进化树上产生“虚假回声”,增加了对哪些分支与分组值得信赖的不确定性。
识别快速变化的位点与隐藏的错误
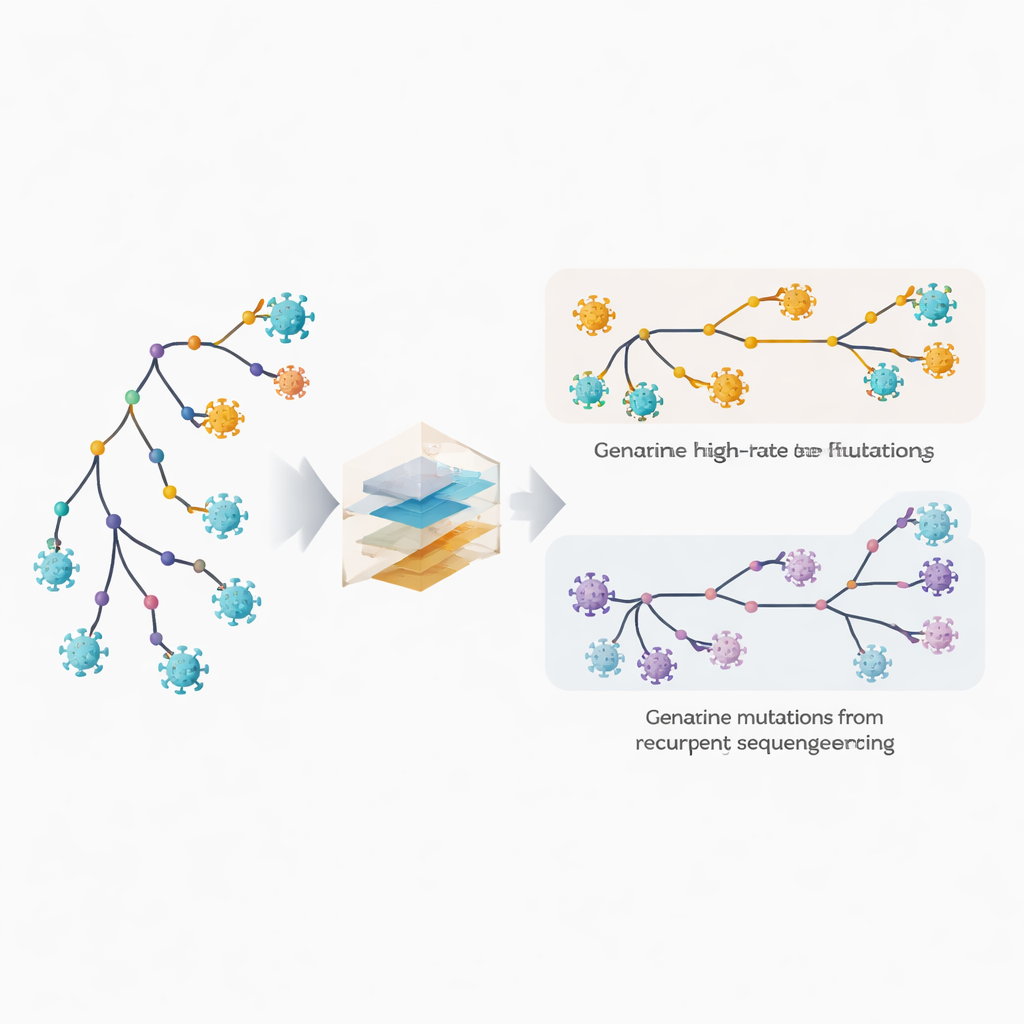
作者扩展了其系统发育软件 MAPLE,采用模型将病毒基因组中的每个位置视为具有独立行为。方法不再假设少数几个平均突变率,而是利用海量基因组为每个位点分别估计速率。同时,它允许每个位点有自身出现重复测序或共识调用错误的概率。关键技巧是比较某一变化在树的深层内部分支(反映更古老的共享事件)与外层末端枝(对应单个基因组)上出现的频率。真实的生物学突变倾向于在内部与末端分支之间分布,而技术性错误主要出现在末端。通过利用这一模式,该方法能够将真正的快速演化与重复错误区分开来。
为拥挤的生命之树设计更快的算法
处理数百万条基因组通常需要巨大的计算能力。为使分析可行,团队重新设计了 MAPLE 在树上存储和更新序列信息的方式。软件不再将每个基因组与单一固定参考进行比较,而是在树内部选择“局部参考”点,并将附近的基因组记录为相对于这些锚点的差异。这种紧凑表示加速了树中远端部分之间的比较。额外的改进包括优化如何向现有树添加新样本、调整支枝长度以及探索替代树形的可能性,并提供在多个处理器核心上并行运行最耗时步骤的选项。
测试方法并清理真实世界数据
为了验证其模型的有效性,作者首先创建了具有已知突变模式并嵌入序列错误的现实模拟 SARS‑CoV‑2 数据集。在这些测试中,新方法恢复了更真实的进化树,并能高精度定位单个错误,尤其是在包含数万条或更多基因组时效果显著。随后他们分析了真实数据,研究了可获得原始读取数据的数百万条 SARS‑CoV‑2 序列。通过比较两种不同的共识构建流程,作者精确定位了反复受工艺伪影影响的特定基因组位点,例如引物结合问题或参考偏倚的调用。这些可疑位点在后续分析中被屏蔽,显示出污染或混合感染迹象的基因组被过滤掉,最终得到超过两百万条高质量序列的整理对齐序列。
更清晰的全球病毒家族树图景
利用清理后的数据集,作者重建了全球 SARS‑CoV‑2 的系统发育树并映射了主要变体之间的关系。他们的树在某些情况下提出的关系与先前公开的树略有不同,通常以更少的突变事件解释并更符合统计模型。该框架还指出了系谱标签可能与潜在遗传历史不一致的地方,标记出可能的重组体或有问题的基因组以便进一步检查。尽管仍存在一些挑战——例如数据稀少时的过拟合,或受严重污染样本的影响——这项工作表明构建更可靠的大流行规模进化树现已可行。对普通读者而言,结论是更好地处理错误与突变热点可带来更清晰的洞见,帮助科学家和卫生机构在未来的疫情中更快、更有把握地做出反应。
引用: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
关键词: SARS-CoV-2 基因组学, 系统发育方法, 测序错误, 突变率变异, 基因组流行病学