Clear Sky Science · zh
高效且精确的机器学习势在材料科学中的空间混合方法
为何更快的原子级模拟很重要
为核聚变、微电子以及结构合金等技术设计更优材料,越来越依赖于追踪原子运动与相互作用的计算模拟。最精确的方法源自量子物理思想,但计算开销极大,仅能处理有限的体系规模和时间尺度。本文介绍了 ML‑MIX,一种方法与软件套件,允许研究者在关键区域保持近乎量子力学的精度,同时在其他区域使用更简单、更廉价的模型。由此可获得显著的加速——通常提高约 4 到 10 倍——且在关键物理预测上不丢失可靠性。
将详细与简化的原子视角混合
工作的核心思想很简单:模拟中并非每个原子都需要相同程度的关注。键被拉伸、断裂或重排的区域——例如缺陷、表面或植入颗粒——受益于现代机器学习原子间势,它们能模仿量子力学的精确性。但远离这些“热点”的原子大多在规则位置附近振动,可以用更简单的模型处理。ML‑MIX 提供了一种在同一模拟盒中组合精确但昂贵模型与更精简“廉价”模型的方法。其做法是定义使用昂贵模型的核心区、在其中对力进行细致混合的缓冲区,以及仅使用廉价描述的外部体相区。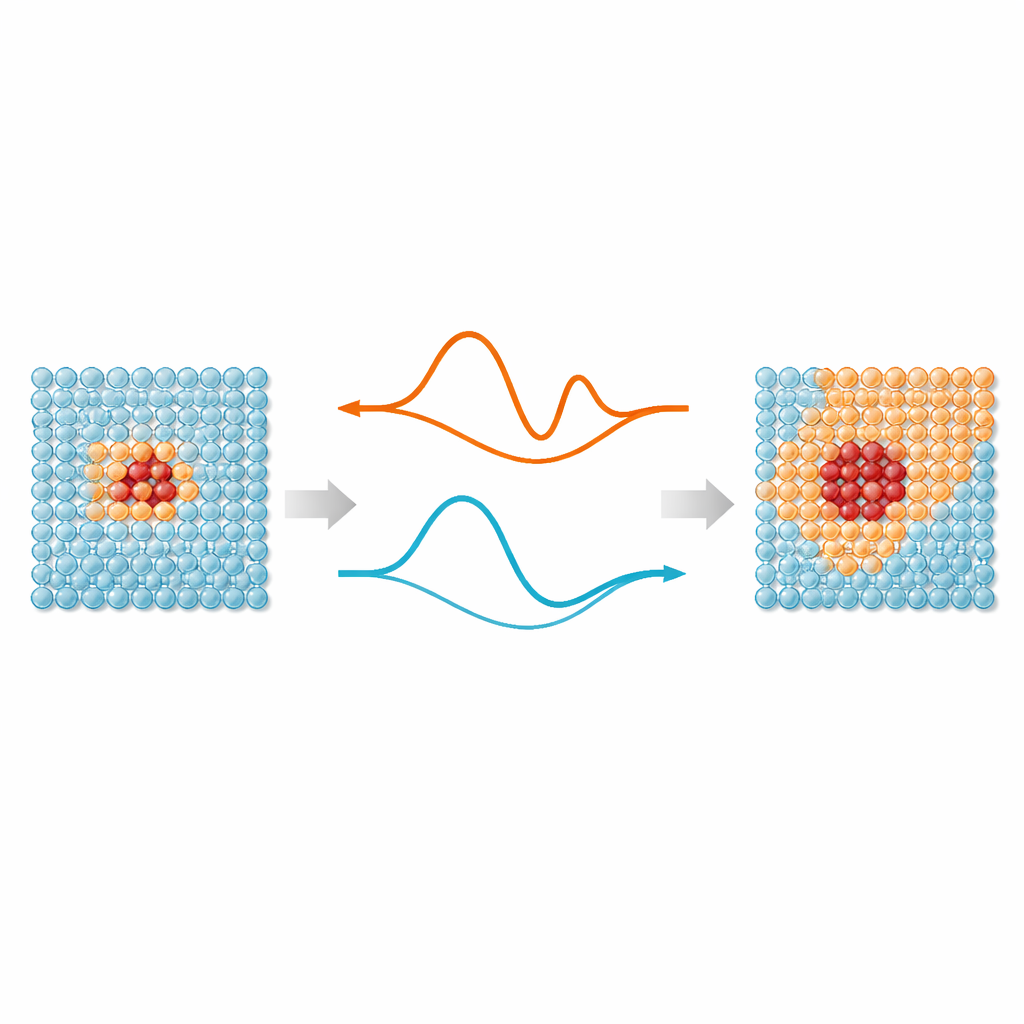
教会廉价模型模仿精确模型
一个关键挑战是确保廉价模型在接触面附近的行为与精确模型一致。作者没有将廉价模型直接拟合到庞大且多样的量子力学数据集上,而是通过在与体相区域相关的特定条件下运行精确模型来生成聚焦的“合成”数据:高温振动和轻微受张的晶体。然后以这些数据为目标拟合廉价模型,同时对弹性常数和晶格常数等基本材料属性施加严格约束。这样的约束拟合确保长程应力与应变在两种模型的边界处平滑匹配,避免产生可能破坏界面附近动力学的人为力。
将方法付诸检验
为验证 ML‑MIX 的有效性,作者在硅、铁和钨体系上进行了一系列测试。一个简单例子是计算硅中空位(一个空的晶格位置)从一个位置迁移到另一个位置的能垒。混合模拟在能量上复现了全昂贵模型计算的结果,误差小于千分之一电子伏特,同时运行速度约快五倍。在更动态的场景中,他们在高温晶体中拉伸一个硅键并测量其平均受力。仅用廉价模型的模拟已经出人意料地接近,但一旦在拉伸键周围加入一个小的昂贵核心,结果在统计上与完全精确的参考无法区分,并在串行运行中实现了高达约 13 倍的加速。
追踪缺陷与颗粒的运动
更现实的测试考察了缺陷在金属中的运动。团队模拟了铁中自间隙缺陷的扩散以及钨内部氦原子的行为。在每种情况下,昂贵模型被限制在围绕缺陷的小规模移动区域,而晶体的其余部分由廉价势处理。所得的扩散系数在统计误差范围内与全精确模拟相匹配,即使仅用廉价模型的模拟会失败。作者随后将该方法推广到更大、具有科学重要性的钨问题——钨是核聚变反应堆的主要候选材料。他们模拟了控制塑性变形的螺位错(线状缺陷)运动,以及氦原子向高温钨表面的植入。在这两种情况下,ML‑MIX 在重现仅昂贵模型结果的同时,将计算成本降低了大约四到十一倍。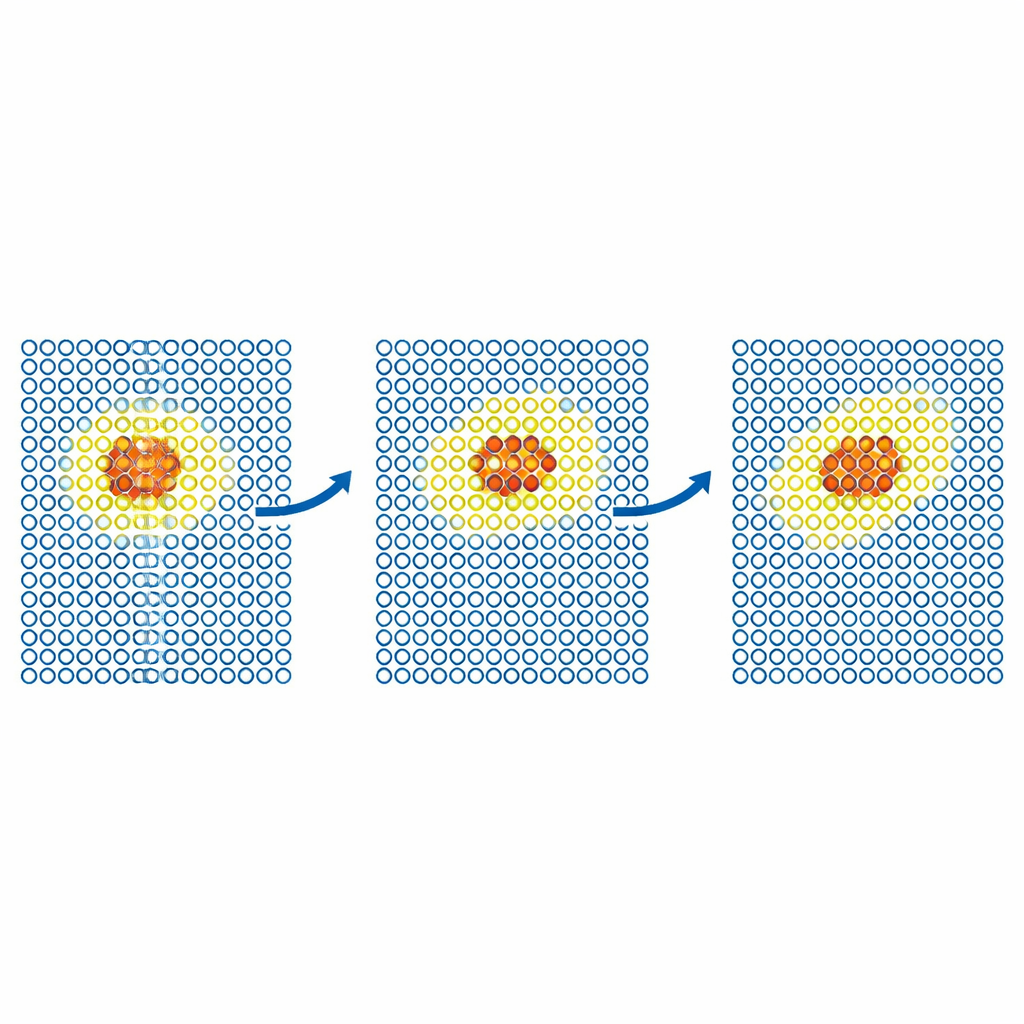
与实验匹配并展望未来
氦注入研究最清晰地展示了这一方法的威力。通过将描述氦—钨相互作用的前沿机器学习模型与用于纯钨的更快势混合,作者得以在图形处理器上模拟更多冲击事件和更大的样本,这在其他情况下不可行。预测的氦原子从表面弹回与植入金属内部的比例,与实验测量在约 80 电子伏特的入射能量范围内相符,而早期模拟难以达到这一点。尽管混合方案并不严格守恒能量并需要温和的恒温方法,但产生的漂移很小且可控。总体而言,ML‑MIX 证明了谨慎结合详细与简化的原子模型能够打破长期存在的精度与尺度之间的矛盾,为在现实环境中常规进行高保真复杂材料模拟打开了大门。
引用: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
关键词: 机器学习原子间势, 多尺度材料模拟, 钨中氦注入, 缺陷与位错, 分子动力学加速